Фукозидоз у ребенка - кратко с точки зрения педиатрии
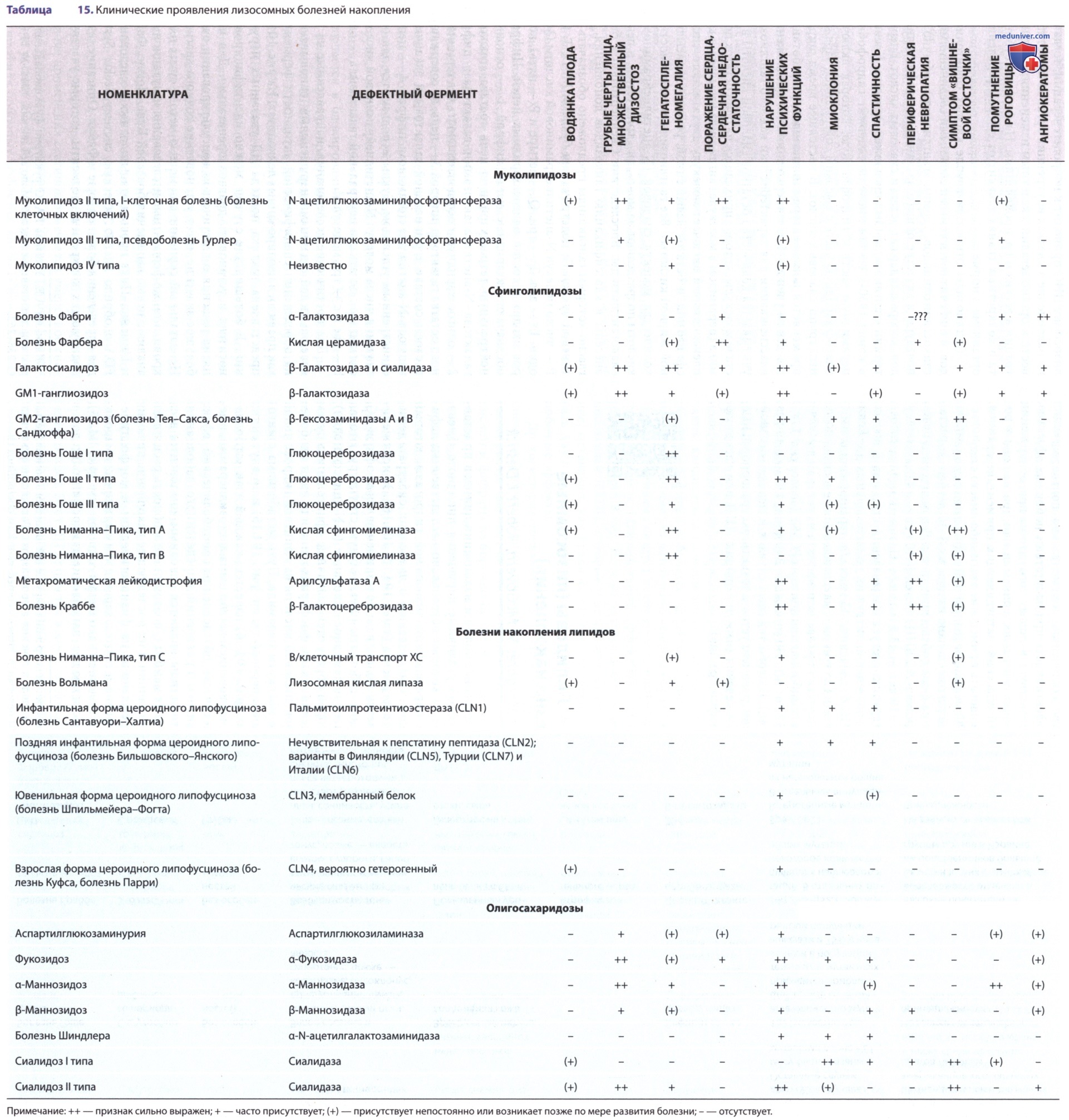
Фукозидоз — это редкое АуР-нарушение, которое развивается в результате дефицита активности α-фукозидазы и накопления фукозосодержащих гликосфинголипидов, гликопротеинов и олигосахаридов в лизосомах печени, ГМ и др. органов (см. табл. 15).
Ген, кодирующий α-фукозидазу, располагается на хромосоме 1 (1p24), специфические мутации известны. Хотя распространенность заболевания не зависит от этнической принадлежности, больше всего пациентов зарегистрировано в Италии и США. Клинические фенотипы широко варьируют.
Самые тяжелые формы проявляются на первом году жизни задержкой развития и соматическими признаками, напоминающими симптомы мукополисахаридоза — выступающими лобными буграми, гепатоспленомегалией, грубыми чертами лица и макроглоссией. Накопление субстрата в ЦНС приводит к неуклонно прогрессирующиему нейродегенеративныму процессу, который завершается летальным исходом в детском возрасте. Для пациентов с более легкими формами характерны наличие ангиокератом и большая продолжительность жизни.
В настоящее время условно выделяют две клинические формы: тяжелую инфантильную (I тип) и более легкую (тип II). Клинические формы представляют собой континуум клинических фенотипов. При типе I дебют заболевания в возрасте 3-18 мес, дети рождаются без особенностей, редко выявляются при рождении пупочные и/или паховые грыжи.
Манифестация заболевания включает прогрессирующую задержку психомоторного и речевого развития, формирование грубых черт лица по типу гаргоилизма, мягкий множественный дизостоз, у больных часто развиваются инфекции верхних и нижних дыхательных путей, отиты. На втором году жизни выявляется задержка роста, умственная отсталость, неврологическая симптоматика (гиперрефлексия, спастический тетрапарез).
У части больных (30%) развивается гепатомегалия или гепатоспленомегалия, могут быть судороги (38% больных детей). Основные скелетные дисплазии включают овоидную или клювовидную деформацию тел позвонков, которая приводит к кифосколизу и тугоподвижности суставов. Ведущим проявлением является поражение НС, формируется умственная отсталость в стадии имбецильности или идиотии, утрачиваются моторные и речевые функции. Летальный исход происходит в возрасте до 10 лет в связи с децеребрационной ригидностью.
При II типе заболевание манифестирует на 1-2-м году, отличительной особенностью считается медленное прогрессирование умственной отсталости и множественного дизостоза, мягкие проявления гаргоилического изменения черт лица. Прогрессирующее отставание по росту имеет место у всех больных, окончательный рост не бывает >135 см. У больных с этой формой встречаются телеангиоэктазии на коже и слизистых, диффузная ангиокератома.
Последняя считается возрастзави-симым признаком: до 10 лет встречается у 1/3 больных, от 10 до 20 лет у 75% и в возрасте >20 лет у 85% больных. Типичные места локализации сосудистых образований — первичное образование в области гениталий и в нижней части живота с последующим распространением по всему телу. У больных выявляют поражение органа зрения, чаще отмечаются извитые и дилатированные вены сетчатки и/или конъюнктивальных сосудов. К редким проявлениям относится тугоухость.
Неврологическая симптоматика отличается выраженным полиморфизмом, отмечается мышечная гипо- и гипертония, судорожный синдром, гиперрефлексия или гипорефлексия. Сохранение речевых и моторных функций после 10-летнего возраста имело место у 53% больных. Продолжительность жизни определяется скоростью прогрессирования неврологических проявлений, в среднем больные погибают после 20 лет, в некоторых случаях живут до 36 лет*.
P.S. * Захарова Е.Ю., Байдакова Б.Г., Михайлова С.В. и др. Лизосомные болезни накопления: руководство для врачей. М.: ГЭОТАР-Медиа, 2021.
Специфической терапии фукозидоза не существует. Диагноз можно установить на основании определения снижения активности а-фукозидазы в лейкоцитах периферической крови или культуре фибробластов. Выявить носителей и установить диагноз пренатально можно путем определения активности фермента или специфических семейных мутаций методом ДНК-анализа.