Лизосомные болезни накопления (липидозы) - кратко с точки зрения педиатрии
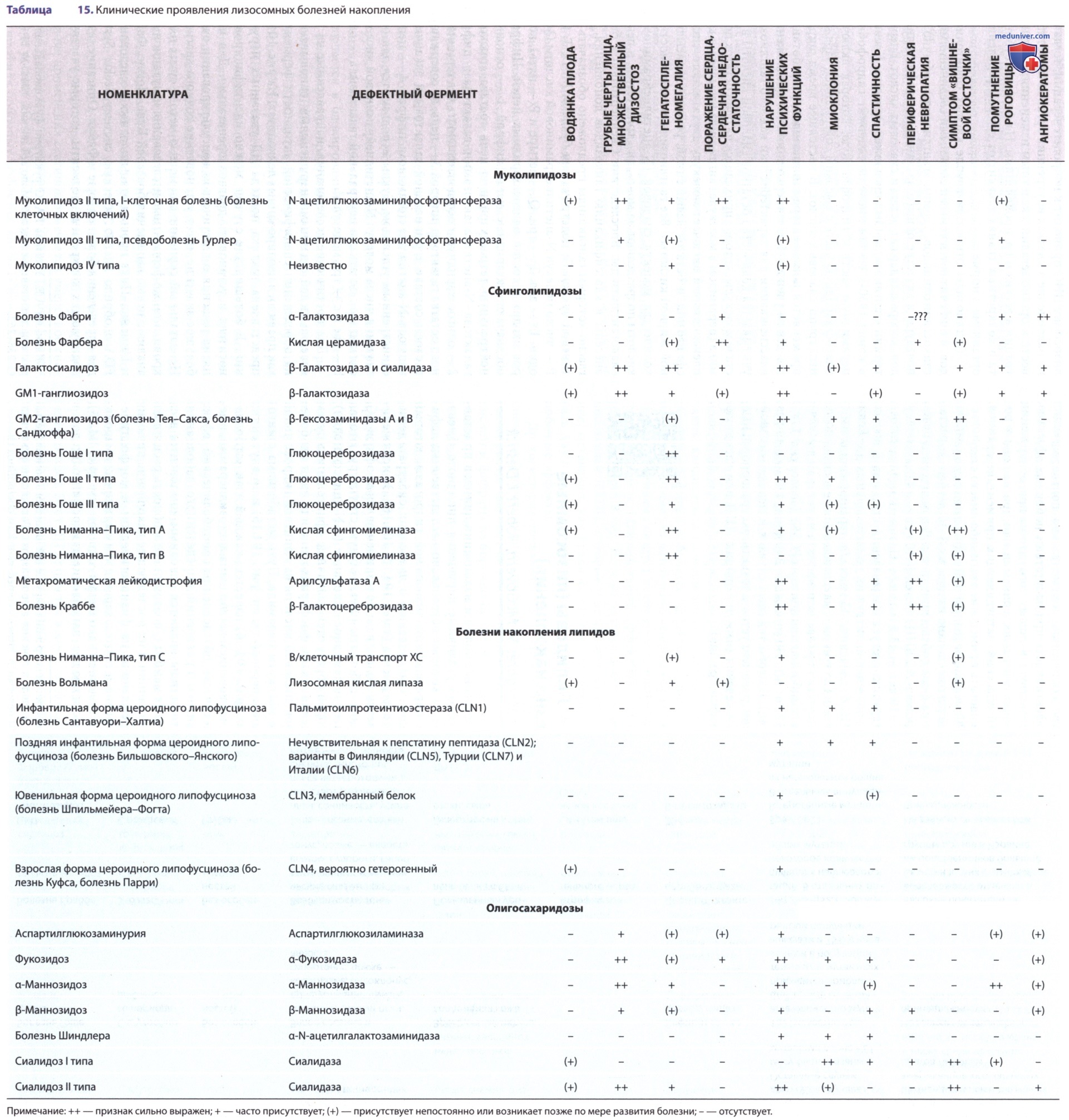
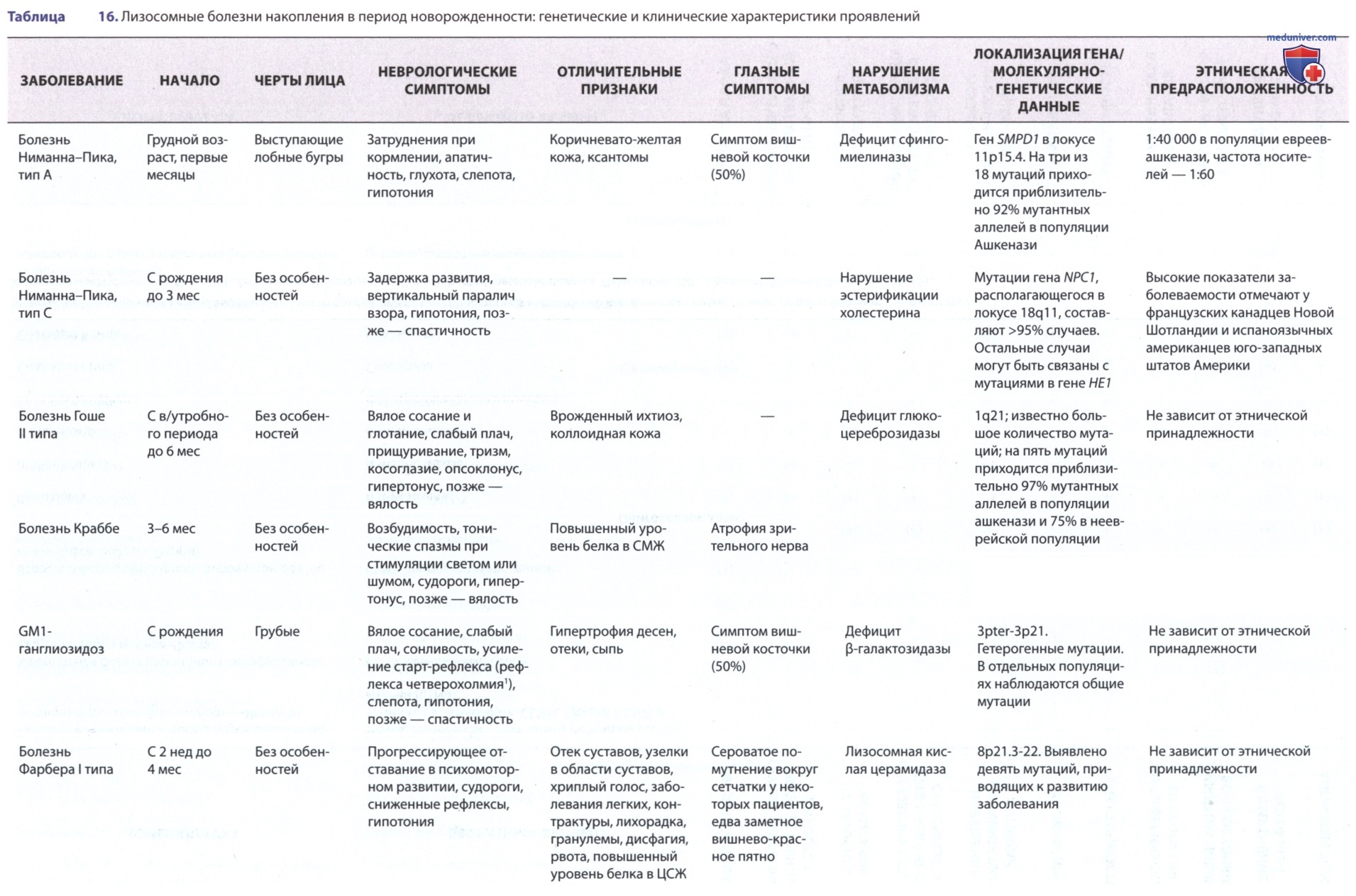
Лизосомные болезни накопления липидов представляют собой различные нарушения липидного обмена, каждое из которых обусловлено наследственным дефицитом определенной лизосомной гидролазы, который приводит к накоплению в лизосомах субстрата дефицитного фермента (табл. 15, 16). За исключением болезни Вольмана и болезни накопления эфиров ХС, липидные субстраты имеют общую структуру, которая включает церамидное основание (2-N-ацилсфингозин), из которой образуются различные сфинголипиды путем замещения гексоз, фосфорилхолина или один или более остатков сиаловой кислоты на концевой гидроксильной группе молекулы церамида.
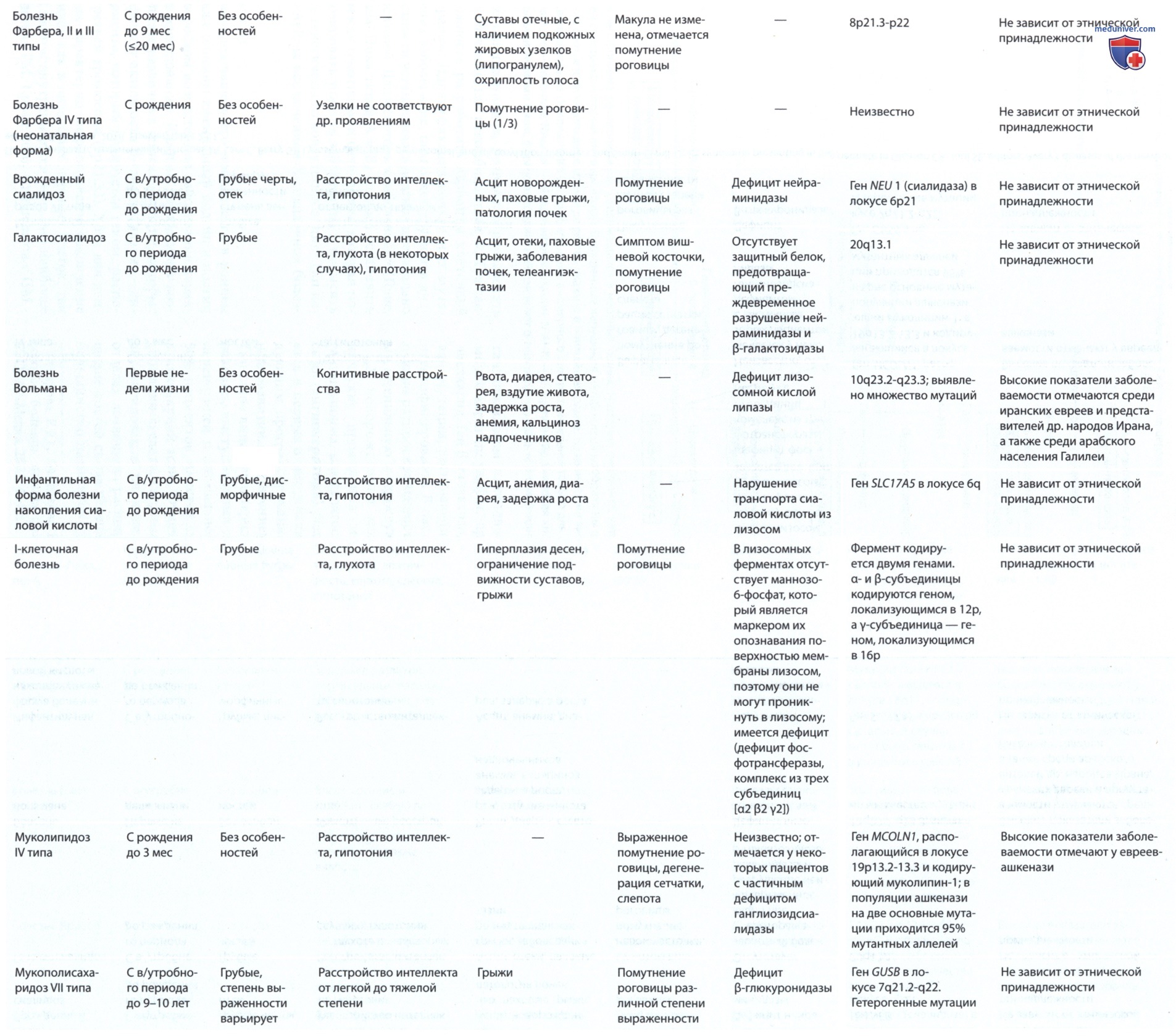
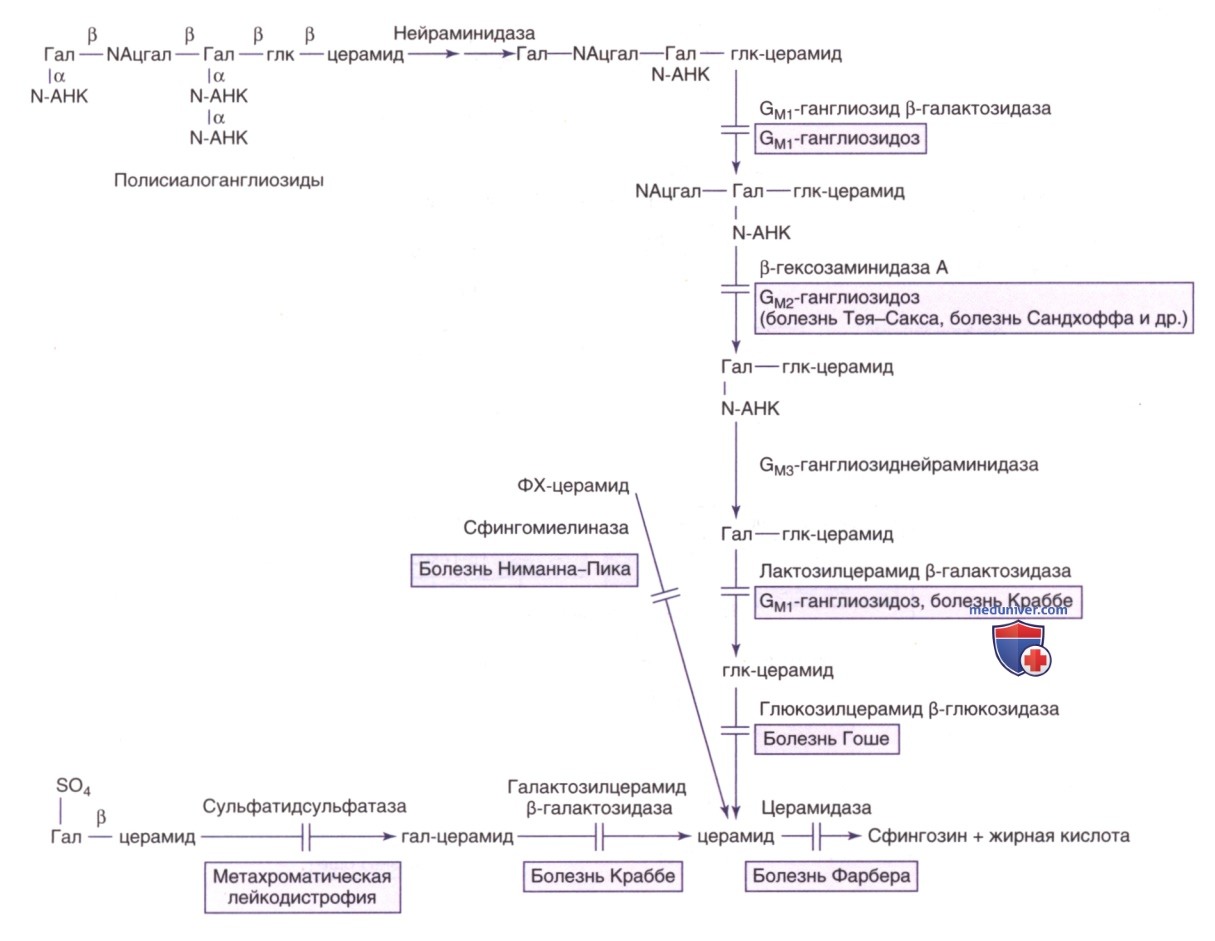
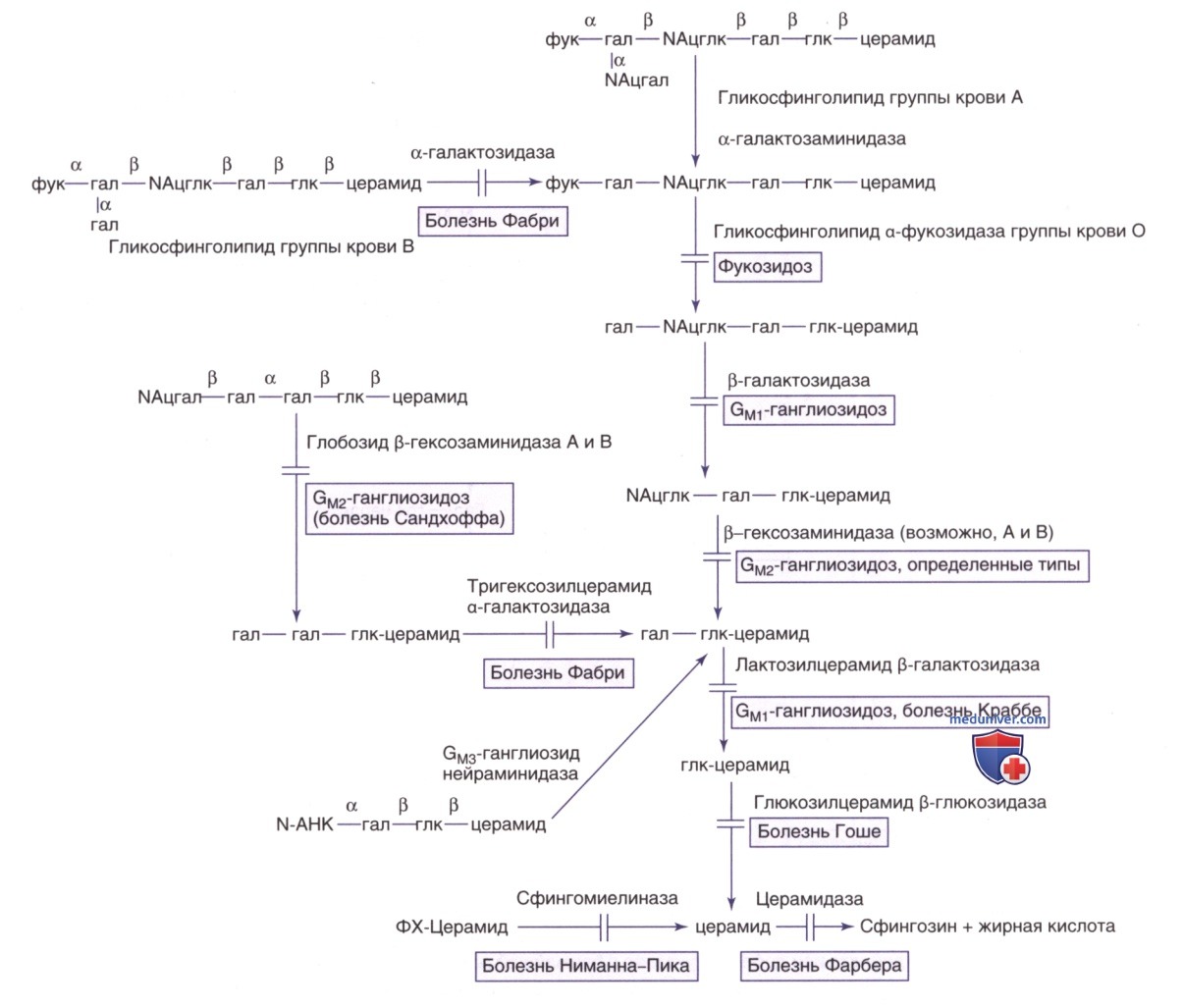
Путь метаболизма сфинголипидов в нервной ткани (рис. 15) и во внутренних органах (рис. 16) известен; каждый этап катаболизма, за исключением катаболизма лактозилцерамида, имеет генетически обусловленный метаболический дефект с развитием проявлений определенного заболевания. Сфинголипиды являются основным компонентом всех клеточных мембран, поэтому неспособность расщеплять эти субстанции и их последующее накопление приводит к повреждениям на физиологическом и морфологическом уровне, а также к характерным клиническим проявлениям болезней накопления липидов (см. рис. 15).
Рисунок 15. Пути метаболизма сфинголипидов, содержащихся в нервных тканях. Название фермента, катализирующего каждую реакцию, дано вместе с названием субстрата, который он гидролизует. Врожденные нарушения метаболизма изображены в виде полосок, пересекающих стрелки реакций, а название соответствующего дефекта или дефектов указано в ближайшей рамке. Названия ганглиозидов представлены в соответствии с номенклатурой Свеннерхольма (Svennerholm L.). Конфигурации аномерного центра приведены только для самого крупного начального соединения. Гал — галактоза; глк — глюкоза; NАцгал — N-ацетилгалактозамин; N-AHK — N-ацетилнейраминовая кислота; ФХ — фосфорилхолин; α и β над горизонтальными линиями в хим. формулах соединений означают а-гликозидную связь и β-гликозидную связь полисиалоганглиозида
Рисунок 16. Пути распада сфинголипидов в тканях внутренних органов, эритроцитах или лейкоцитах. См. также пояснения к рис. 15. Фук — фукоза; NАцглк — N-ацетилглюкозамин
Прогрессирующее накопление гликосфинголипидов в лизосомах ЦНС приводит к нейродегенерации, тогда как их накопление в клетках внутренних органов может привести к органомегалии, скелетным изменениям, инфильтрации в легких и др. проявлениям. Накопление субстрата в конкретных тканях зависит от его распределения в организме в норме.
Ганглиозиды относят к группе кислых сфинголипидов, и они считаются самыми сложными соединениями из гликосфинголипидов. Термин «ганглиозиды» обычно используют в качестве общего названия гликосфинголипидов, которые содержат остатки сиаловых кислот. Впервые термин «ганглиозиды» был предложен Эрнестом Кленком в конце 1930-х гг.
Ганглиозиды содержат гидрофобную церамидную часть и гидрофильную, которая включает олигосахаридную часть с высоким содержанием заряженных групп сиаловых кислот. В настоящее время широко применяется номенклатура ганглиозидов, разработанная шведским ученым Л. Свеннерхольмом. По предложенной классификации для характеристики каждого ганглиозида учитывается количество остатков сиаловой кислоты в составе молекулы, которое приходится на один остаток церамида (церамидом в молекуле ганглиозида называется комплекс, который включает молекулу двухатомного спирта сфингозина и остаток насыщенной, реже — ненасыщенной жирной кислоты).
Все ганглиозиды в зависимости от количества молекул сиаловой (N-ацетилнейраминовой) кислоты подразделяют на моно-, ди-, три-, тетра-, и пентасиалоганглиозиды.
Впоследствии Свеннерхольмом были внесены в классификацию гликозидов дополнения, согласно которым ганглиозиды предложено обозначать латинской буквой G; подстрочными буквами М, D, Т, Q и Р — число молекул N-ацетилнейраминовой кислоты (М — один, D — два, Т — три, Q — четыре и Р — пять), которое входит в состав олигосахаридных цепей. Цифрами обозначают сахаридные цепи: цифрой 1 — основную нейтральную тетрасахаридную цепь состава «глюкоза-галактоза-N-ацетилгалактозамин-галактоза»; цифрой 2 — олигосахаридную последовательность без терминального остатка галактозы; цифрой 3 — цепь, не содержащую в своем составе терминальной галактозы и заканчивающуюся N-ацетилгалактозамином; цифрой 4 — цепь с единственным углеводом.
Буквы используются для обозначения числа молекул N-ацетилнейраминовой кислоты, связанных с остатками центральной галактозы: а — одна, b — две и с — три молекулы*.
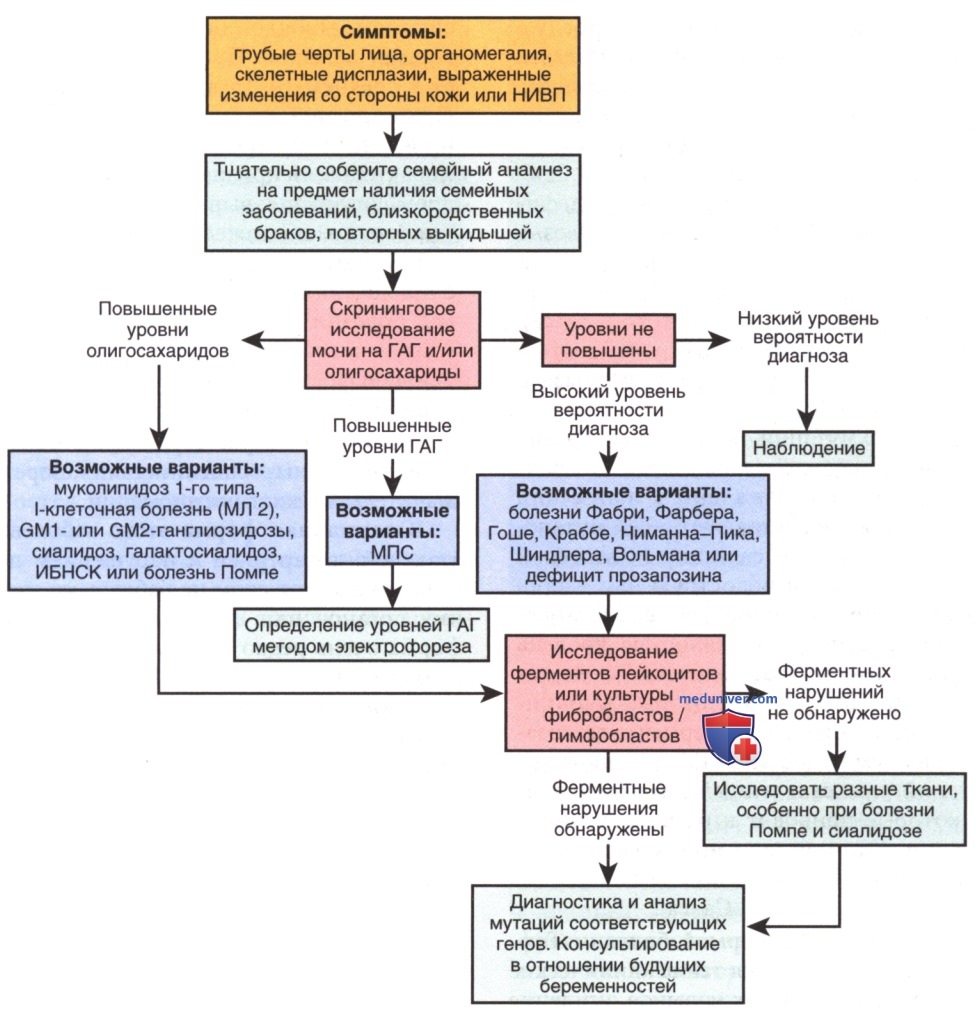
Диагностические исследования, направленные на выявление людей с данными нарушениями, основываются на определении специфической активности ферментов, как правило, в изолированных лейкоцитах. На рис. 17 представлена тактика подхода дифференциальной диагностики (ДД) указанных нарушений. В большинстве случаев можно идентифицировать носителя и установить диагноз пренатально.
Рисунок 17. Алгоритм клинического обследования детей грудного возраста с подозрением на лизосомные болезни накопления. ГАГ — гликозаминогликаны; ИБНСК — инфантильная форма болезни накопления сиаловой кислоты; НИВП — неиммунная водянка плода
Для принятия решения о генетическом консультировании очень большое значение имеет установление точного диагноза. Неонатальный скрининг с помощью метода сухих пятен крови, а также ферментные исследования и мутационный анализ с целью выявления болезней Гоше, Помпе, Фабри и Ниманна-Пика прошли этап пилотных исследований. FDA одобрило диагностическую систему Seeker System для определения болезней Гоше и Фабри.
Описание генов, кодирующих конкретные ферменты, которые участвуют в метаболизме сфинголипидов, позволило разработать такие варианты лечения, как рекомбинантная ФЗТ, а также дает возможность разработать варианты клеточной или генной терапии. Идентификация конкретных мутаций, вызывающих заболевание, позволяет улучшить диагностику, в частности устанавливать диагноз пренатально и выявлять носителей. Для некоторых заболеваний (Гоше, Фабри, Ниманна-Пика типов А и В) удалось установить корреляцию между генотипом и фенотипом, которая позволяет прогнозировать тяжесть заболевания и повысить эффективность генетического консультирования. Тип наследования — АуР, за исключением Х-сцепленной болезни Фабри.