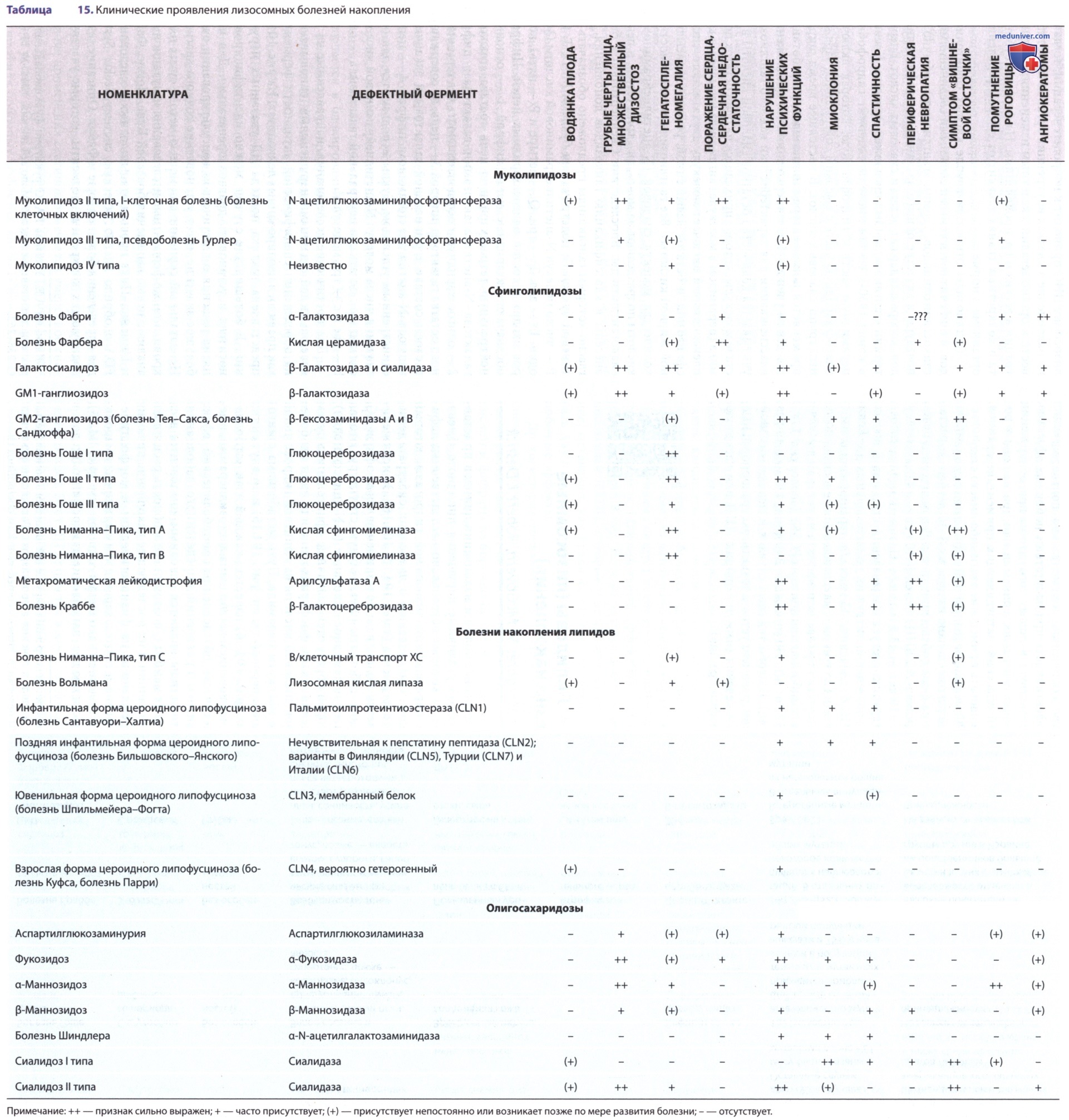
Болезнь Фабри — Х-сцепленное врожденное нарушение метаболизма гликосфинголипидов, обусловленное отсутствием или выраженным дефицитом активности α-галактозидазы А (α-гал-А). Имеется два основных фенотипа.
В КР РФ выделяют две клинические формы болезни Фабри, которые соответствуют основным фенотипам: классическую (дебют в любом возрасте, мультисистемное поражение) и неклассическую (позднее начало, изолированное поражение одной системы органов: ГМ, сердца или почек)*.
P.S. КР «Болезнь Фабри», год утверждения: 2019; возрастная категория: взрослые, дети. Разработчик клинической рекомендации: Ассоциация медицинских генетиков, Союз педиатров России.
У пациентов мужского пола с классическим фенотипом (формой) заболевание проявляется в детском возрасте ангиокератомами (телеангиоэктатическими кожными очагами), гипогидрозом, помутнением роговицы и хрусталика, а также болезненными акропарестезиями. С возрастом развиваются почечная недостаточность, заболевания сердца и инсульт (см. табл. 15). Классический фенотип появляется в результате отсутствия активности α-гал-А, а его распространенность составляет 1:40 000 мужского населения. Фенотип с поздним началом (неклассическая форма) возникает у мужчин с остаточной активностью α-гал-А и проявляется с четвертого по восьмое десятилетие жизни заболеваниями сердца и почечной недостаточностью.
Этот фенотип распространен больше, чем классический. У гетерозиготных женщин с классическим фенотипом заболевание может протекать бессимптомно или так же тяжело, как у мужчин. Такая вариабельность является результатом случайной инактивации Х-хромосомы. Дефицит фермента связан с мутациями в гене, кодирующем α-гал-А и располагающемся на длинном плече Х-хромосомы (Xq22).
Учитывая гетерогенность клинического фенотипа болезни Фабри, частота этой болезни в разл. странах варьирует в широких пределах (от 1:117 000 до 1:476 000 живых новорожденных). Частота новых случаев, оцененная на выборках новорожденных мальчиков, составила в Италии 1:3100, а на Тайване 1:2400. Вероятно, что более распространенным является легкое, атипичное течение болезни с признаками поражения одного органа.
При проведении скрининга групп высокого риска у мужчин и женщин с ранним инсультом доля болезни Фабри составила 4,2 и 2,15%, соответственно, с гипертрофией ЛЖ неясного происхождения — 0,9-3,9 и 1,1-11,8%, с терминальной почечной недостаточностью — 0,33 и 0,10%.
Первичным биохим. дефектом при болезни Фабри является недостаточность фермента α-гал-А, который отщепляет терминальный остаток α-галактозы олигосахаридной цепи нейтральных гликосфинголипидов.
Дефект фермента приводит к системному накоплению нейтральных гликосфинголипидов, прежде всего глоботриаозилцерамида, особенно в плазме и лизосомах сосудистого эндотелия, гладкомышечных клеток, кардиомиоцитов и подоцитов почек. Прогрессирующее отложение гликосфинголипидов у мужчин с классическим фенотипом болезни Фабри приводит к окклюзии мелких сосудов и ишемии, что вызывает развитие основных проявлений заболевания. Описаны кДНК и геномные последовательности, кодирующие α-гал-А. Выявлено более 900 различных мутаций в гене, кодирующем α-гал-А, вследствие которых развивается данная лизосомная болезнь накопления.
К настоящему времени идентифицировано >600 вариантов в гене GLA, в т.ч. ок. 500 патогенных мутаций, изменяющих свойства и стабильность фермента α-гал-А.
Гемизиготные мужчины часто имеют характерный внешний вид, напоминающий больных с акромегалией: выступающие супраорбитальные дуги и лобные бугры, выступающая нижняя челюсть, увеличенные губы, запавшая переносица.
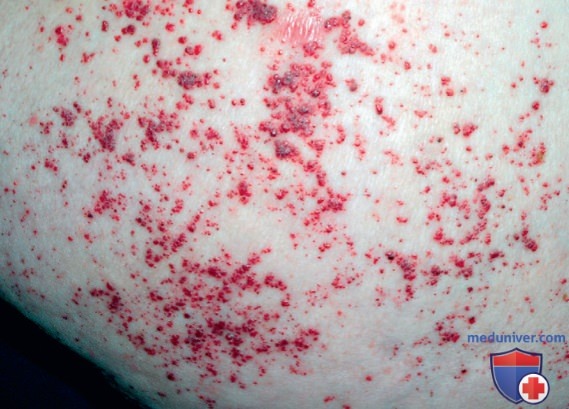
Ангиокератомы, как правило, возникают в детском возрасте, их выявление способствует раннему установлению диагноза (рис. ниже).
Типичные ангиокератомы. Ангиокератомы имеют довольно большие размеры и легко распознаются, однако их легко не заметить, когда они представлены только несколькими элементами или ограничены лишь областью гениталий или пупочной областью
Ангиокератомы образуются из скоплений кровеносных сосудов в верхних слоях дермы и утолщения эпидермиса в виде поверхностного гиперкератоза. Очаги на начальной стадии плоские, более светлого цвета и легче поддаются сжатию.
С возрастом их количество возрастает, и они увеличиваются в размерах от едва видимого до нескольких миллиметров в диаметре. Элементы точечные, цвет варьирует от темно-красного до сине-черного, они плоские или слегка возвышаются над поверхностью кожи. Они не бледнеют при надавливании. У более крупных элементов может наблюдаться гиперкератоз. Типично наиболее плотное расположение элементов в области между пупком и коленными суставами, в паховой области, однако элементы могут возникать в любом месте, в т.ч. на слизистой оболочке полости рта, конъюнктиве, ладонях. Ангиокератомы часто появляются в области тазобедренных суставов, бедер, ягодиц, пупка, нижней части живота, мошонки и головки пениса, причем отмечается тенденция к симметричности. Описаны варианты без поражения кожи.
Активность потовых желез, как правило, снижена или отсутствует. Снижение или полное отсутствие потоотделения обусловлено накоплением глоботриаозилцерамида в потовых железах и в стенках кровоснабжающих их сосудов с поражением вегетативных нервных волокон (вегетативный вариант полиневропатии тонких волокон). В редких случаях наблюдается гипергидроз*.
P.S. * КР «Болезнь Фабри», год утверждения: 2019; возрастная категория: взрослые, дети. Разработчик клинической рекомендации: Ассоциация медицинских генетиков, Союз педиатров России.
У больных мужчин, а также у 90% гетерозигот из семей с классическим фенотипом наблюдают помутнение роговицы и характерное поражение хрусталика. У многих пациентов наблюдается извитость сосудов конъюнктивы и сетчатки, которая возникает в результате системного поражения сосудов.
У пациентов с болезнью Фабри в 60-80% случаев наблюдается хроническая выраженная, изнуряющая невропатическая боль в конечностях. Типичны такие жалобы, как длительная жгучая боль, стреляющая пронзающая боль, боль, подобная электрическому разряду. Акропарестезии представляют собой спонтанные неприятные неболевые ощущения по типу покалывания иголками, ползания мурашек в кистях и стопах. Отмечается аллодиния — боль в ответ на неболевой стимул, гиперпатия — чрезмерная болевая реакция на болевой стимул. Она может быть как механической, т.е. боль на прикосновение кисточкой, ваткой, так и температурной при термическом воздействии, при повышении температуры окружающей среды. Некоторые пациенты также отмечают леденящую боль в области ладоней на морозе.
Кризы Фабри продолжаются от нескольких часов до нескольких дней, состоят из мучительной, жгучей боли в кистях, стопах и проксимальных отделах конечностей и обычно связаны с физической нагрузкой, усталостью, лихорадкой или сочетанием этих факторов.
Эти эпизоды могут возникнуть у лиц обоих полов и начаться уже с двух лет. При кризе Фабри боль приобретает более мучительный характер, может иррадиировать из дистальных отделов конечностей в проксимальные, длится от нескольких секунд до нескольких недель и не снимается наркотическими анальгетиками.
Механизмы невропатической боли при болезни Фабри полностью в настоящее время не изучены. Предположительно боль связана со структурными повреждениями тонких нервных волокон в результате накопления глоботриаозилцерамида в аксонах нервов, ганглиях задних корешков и в vasa nervorum (лат. «сосуды нервов»). Это болевой вариант полиневропатии тонких волокон, периферическая невропатическая боль. Известно также о кальцификации таламуса при болезни Фабри (центральная невропатическая боль при повреждении таламуса).
В третьем или четвертом десятилетии жизни приступы болезненной акропарестезии обычно возникают реже, однако у некоторых мужчин они становятся более частыми и тяжелыми. Приступы боли в животе или боку могут стимулировать аппендицит или почечную колику. Боли при акропарестезиях могут трактоваться как проявления др. заболеваний (табл. 17).
Основные симптомы заболевания у мужчин с классическим фенотипом развиваются в результате прогрессирующего поражения сосудистой системы. Первые симптомы поражения почек — микроальбуминурия и протеинурия — чаще возникнуть в подростковом и взрослом возрасте, но могут возникнуть и на первом десятилетии жизни. Поражение почек также часто дебютирует в 3-5 лет, однако клинически оно никак не проявляется. Первым признаком дисфункции почек, часто имеющем место у детей и подростков, является эпизодическая микропротеинурия и микроальбуминурия (экскреция альбумина с мочой от 30 до 300 мг/сут).
При классическом фенотипе на ранней стадии заболевания в осадке мочи появляются цилиндры, эритроциты и липидные включения с двойным лучепреломлением в виде мальтийского креста. Протеинурия, изостенурия и постепенное ухудшение функции почек, а также азотемия при классическом фенотипе развиваются на втором-четвертом десятилетии жизни, а при форме с поздним началом — на четвертом-восьмом десятилетии.
С возрастом протеинурия постепенно нарастает и может достигать нефротического уровня, хотя нефротический синдром развивается редко. Часто отмечаются канальцевые нарушения. Постепенно снижаются функции почек и формируется ХПН. У пациентов с неклассической болезнью Фабри возможно изолированное поражение почек. В таких случаях диагноз может быть установлен на основании исследования почечного биоптата. Могут возникать симптомы со стороны ССС — аритмии, ГКМП и СН. Поражение сердца относят к числу распространенных и прогностически неблагоприятных проявлений болезни Фабри. Заболевания сердца стоят на первом месте среди причин смерти у пациентов с болезнью Фабри (М/Ж 34:57%).
Характерным признаком поражения сердца при болезни Фабри является гипертрофия ЛЖ, которая м.б. выявлена с помощью ЭКГ, ЭхоКГ и МРТ. Проявления со стороны сердца обусловлены не только отложением гликосфинголипидов в миоцитах и проводящей ткани, но и развитием гипертрофии миокарда и фиброза.
Гипертрофия и фиброз миокарда часто сопровождаются диастолической дисфункцией ЛЖ. Для оценки функции сердца применяют тканевую допплерэхокардиографию (допплер-ЭхоКГ), результаты которой важны в диагностике бессимптомного поражения сердца. У подростков при болезни Фабри бывают периодические подъемы или падения АД или бессимптомные варианты аритмий по данным ЭКГ. Крайне сложно дифференцировать ранние проявления этого заболевания от возрастных вегетативных нарушений. Наиболее частым отклонением со стороны клапанов сердца является недостаточность митрального клапана. Цереброваскулярные проявления, в т.ч. ТИА и инсульт, возникают вторично на фоне аритмий, а также множественного поражения малых сосудов.
При этом заболевании высокий риск развития ТИА и ишемических/геморрагических инсультов уже в молодом возрасте. Частота инсульта М/Ж 6,9:4,3%. У большинства больных инсульт развивается в возрасте от 20 до 50 лет, в т.ч. у каждого пятого из этих больных — в возрасте до 30 лет.
Геморрагический инсульт у мужчин встречается чаще, чем у женщин. У 2/3 больных инсульт является следствием атипичного течения болезни с изолированным поражением сосудов ГМ. В 50% болезнь Фабри дебютирует с мозгового инсульта (неклассическая форма), поэтому эту болезнь рекомендуется предполагать у всех больных с ранним развитием инсульта даже при отсутствии очевидных причин и факторов риска. При хронической ишемии ГМ может развиваться сосудистая деменция, для которой характерно снижение памяти и поведенческие нарушения.
Глазные проявления болезни Фабри включают типичный симптом: помутнение роговицы в виде завитков (70-90% больных). Редко возможно развитие задней субкапсулярной катаракты и поражение сосудов сетчатки. Специфическое «мутовчатое» помутнение роговицы (так называемая «воронкообразная» кератопатия, или cornea verticillata) — частый симптом у детей и подростков при болезни Фабри. Подобные специфические изменения выявляются уже в возрасте 4-5 лет, что позволяет предположить наличие болезни Фабри на ранней стадии. Такие изменения, как помутнение хрусталика в виде радиальной задней субкапсулярной катаракты — катаракты Фабри и двусторонней передней капсулярной и подкапсулярной катаракты, редко встречаются у детей и являются патогномоничными симптомами у взрослых больных.
У пациентов с болезнью Фабри могут быть слуховые и вестибулярные нарушения: снижение слуха, шум в ушах, головокружение. К более ранним симптомам, возникающим в детском и подростковом возрасте, относятся шум (звон) в ушах (одно- или двусторонний) и головокружение, к которым с возрастом присоединяется нейросенсорная тугоухость.
К признакам заболевания относят также хронический бронхит и одышку, лимфедему в области голеней без гипопротеинемии, эпизодическую диарею, остеопороз, задержку роста и отсроченный пубертат. Поражение лимфатических сосудов проявляется в подростковом или взрослом возрасте и при отсутствии лечения может привести к выраженным трофическим изменениям с изъязвлениями кожного покрова нижних конечностей и осложняться рожистым воспалением и сепсисом.
а) Особенности клинической картины в детском возрасте. Первые клинические проявления чаще всего возникают в подростковом и юношеском возрасте, хотя иногда признаки заболевания могут дебютировать и в 2-4 года. Ранние проявления болезни Фабри могут быть неспецифическими, что значительно затрудняет диагностику. Первыми проявлениями у детей могут быть боль, дискомфорт в животе, неоформленный стул, сухость кожных покровов, плохая переносимость жары, иногда приводящая к развитию обмороков, эпизодические боли в стопах или кистях. Если боли в руках дебютируют в раннем возрасте, их очень сложно верифицировать, т.к. эпизоды болей выглядят как истерика, психомоторное возбуждение.
Косвенным признаком болей в пользу периферической акропарестезии при болезни Фабри является то, что ребенок успокаивается в прохладной воде (бассейн), или облегчение, если больной ложится на холодную поверхность (напр., кафельный пол).
В дальнейшем жалобы на боли в конечностях становятся более частыми и постепенно приобретают типичные для болезни Фабри проявления. Цереброваскулярные нарушения у детей с риском развития инсульта являются редкими проявлениями, хотя в литературных данных есть информация о случаях преходящих нарушений мозгового кровообращения с 12 лет.
Гастроэнтерологические проявления болезни м.б. одним из первых симптомов заболевания в детском и юношеском возрасте и возникать на первом десятилетии жизни, но они являются косвенными маркерами болезни Фабри в силу малой специфичности без сочетания с др. симптомами. Наиболее часто наблюдаются схваткообразные боли в животе, вздутие живота, неустойчивый стул, тошнота, рвота снижение аппетита и дефицит МТ.
К редко встречающимся и менее специфичным симптомам болезни Фабри у детей относятся особенности внешности (изменения по типу акромегалии), нарушения дыхания, анемия, скелетные аномалии (деформация дистальных отделов межфаланговых суставов пальцев рук с нарушением их подвижности, утолщение концевых фаланг пальцев по типу «барабанных палочек»), остеопения, остеопороз, трещины и эритематозные изменения грибовидных сосочков на дорсальной поверхности языка; глоссит, гранулематозный хейлит, гипотиреоз, задержка полового развития, приапизм*.
P.S. * КР «Болезнь Фабри», год утверждения: 2019; возрастная категория: взрослые, дети. Разработчик клинической рекомендации: Ассоциация медицинских генетиков, Союз педиатров России
Летальные исходы чаще всего связаны с почечной недостаточностью, заболеваниями сердца или инсультом. До начала применения гемодиализа и трансплантации почек летальный исход у больных мужчин наступал в среднем в возрасте ок. 40 лет. У пациентов с поздним фенотипом с остаточной активностью а-гал-А имеют место сердечные и/или почечные заболевания. Проявления со стороны сердца включают гипертрофию ЛЖ и МЖП, а также отклонения ЭКГ-показателей, соответствующие кардиомиопатии. На фоне ГКМП может возникать желудочковая тахикардия, приводящая к летальному исходу.
б) Диагностика болезни Фабри. Диагноз болезни Фабри устанавливается на основании совокупности анамнестических и клинических данных, результатов лабораторного исследования (биохимического и молекулярно-генетических исследований).
Классический фенотип (форму) болезни Фабри предполагают при наличии типичных проявлений системного заболевания с поражением почек, сердца, кожи, НС, ЖКТ, органа зрения.
У мужчин с классической формой болезни Фабри диагноз прежде всего устанавливается на основании наличия в анамнезе приступов болезненной парестезии, гипогидроза, характерного поражения кожи, а также помутнения роговицы и поражения хрусталика.
Болевой синдром при болезни Фабри часто сопровождается субфебрильной лихорадкой и повышенной СОЭ, что часто приводит к установлению ложных диагнозов: РА, ревматической лихорадки, артритов, эритромелалгии, синдрома Рейно.
Необходимо проводить ДД поражений кожи с доброкачественными ангиокератомами мошонки (болезнь Фордайса) или с ограниченной ангиокератомой. При фукозидозе, аспартилглюкозаминурии, GM1-ганглиозидозе с поздним началом, галактосиалидозе, дефиците α-N-ацетилгалактозаминидазы и сиалидозе отмечают ангиокератомы, аналогичные тем, которые наблюдают при болезни Фабри.
Выявление ангиокератом является важным диагностическим ключом при болезни Фабри. При кризах Фабри, сопровождающихся лихорадкой и болями, ангиокератомы могут быть приняты за петехиальную сыпь, что приводит к установлению ошибочного диагноза менингита. ДД следует также проводить с наследственной геморрагической телангиэктазией Рендю-Ослера-Вебера.
При неклассической форме поражение сердца или почек м.б. единственным проявлением болезни Фабри. У таких пациентов отсутствуют какие-либо симптомы заболевания в детском возрасте. Диагностировать атипичные варианты болезни Фабри можно только путем скрининга групп риска. Предполагать болезнь Фабри следует у всех пациентов с гипертрофией миокарда ЛЖ неясного происхождения и инсультом, развившимся в молодом возрасте (до 40 лет). При изолированном поражении почек мочевой синдром может ошибочно интерпретироваться как хронический латентный гломерулонефрит или интерстициальный нефрит. Пациентов с поздней формой выявляют среди тех, кто проходит процедуру гемодиализа, а также среди пациентов с ГКМП или среди перенесших криптогенные инсульты.
Симптомы поражения ССС при болезни Фабри сходны с кардиомегалией или аритмией неясного генеза. При позднем начале заболевания ранние классические проявления, описанные выше, отсутствуют.
в) Данные физикального обследования. Выраженность клинических проявлений болезни Фабри может варьировать в зависимости от возраста, при физикальном осмотре могут проявляться следующие признаки заболевания:
• грубые черты лица;
• ангиокератома (внутренняя поверхность бедер, кисти, живот, слизистые ротовой полости);
• признаки увеличения сердца;
• признаки периферического болевого синдрома в рамках дистальной полиневропатии с преимущественным поражением тонких волокон;
• признаки перенесенного инсульта (парезы, нарушения речи).
Диагноз классической формы болезни Фабри и формы с поздним началом подтверждают биохимически по значительному снижению активности α-гал-А в плазме крови, лейкоцитах, культуре фибробластов или лимфобластов. Рекомендовано определение активности α-гал-А в пятнах крови, высушенных на фильтровальной бумаге или в лейкоцитах крови всем пациентам мужского пола (в т.ч. и на доклинической стадии) для подтверждения диагноза. У мужчин сниженная активность α-гал-А является достаточно информативным признаком болезни Фабри. Однако у трети женщин активность α-гал-А м.б. в норме, поэтому при проведении ферментного теста женщинам это нужно учитывать.
Рекомендовано проведение молекулярно-генетического исследования для всех пациентов со сниженной активностью фермента а-гал-А и для лиц женского пола при клиническом подозрении на болезнь Фабри при наличии родственников с болезнью Фабри в родословной: выявление мутаций в гене GLA, кодирующем α-гал-А, для подтверждения диагноза на молекулярно-генетическом уровне у мужчин со сниженной активностью фермента и у женщин с клиническими признаками болезни Фабри или родственников пробанда по материнской линии. При получении положительного или сомнительного результата ферментного теста у мужчин и у женщин проводится подтверждающий молекулярно-генетический анализ гена GLA.
Широкое распространение аллелей псевдодефицита (напр., p.D313Y), приводящих к сниженной активности α-гал-А в сухом пятне крови и иногда пониженной — в лейкоцитах и плазме крови, обусловливает обязательное включение молекулярно-генетического анализа в лабораторную диагностику болезни Фабри.
Выявление семейной мутации гена GLA делает возможным обследование родственников пробанда, выявление гетерозиготных носительниц болезни Фабри, а также проведение пренатальной и преимплантационной диагностики. Большинство мутаций у пациентов с болезнью Фабри могут быть выявлены с помощью секвенирования всех экзонов и приэкзонных участков интронов по Сэнгеру, но в небольшом проценте случаев мутацию с применением стандартных методов обнаружить не удается.
Специфическую мутацию в гене, кодирующем α-гал-А, можно определить путем секвенирования генома. У гетерозиготных женщин из семей с классическим фенотипом могут наблюдаться помутнение роговицы, изолированное поражение кожи и промежуточные показатели активности α-гал-А в плазме крови или клетках. В редких случаях у гетерозиготных женщин заболевание проявляется так же тяжело, как у больных мужчин. Однако у женщин с бессимптомным течением заболевания из группы риска, которые принадлежат к семьям с классической формой и формой с поздним началом, оптимальным методом выявления специфической семейной мутации является прямой тип ДНК-диагностики.
У плодов мужского пола пренатальный диагноз можно установить по дефициту активности α-гал-А и обнаружению специфической семейной мутации гена в ворсинках хориона в первом триместре или в культуре амниоцитов, полученных путем амниоцентеза во втором триместре беременности. Болезнь Фабри можно выявить при проведении неонатального скрининга.
Рекомендовано всем пациентам с клинической картиной болезни Фабри определение концентрации глоботриаозилсфингозина (Lyso-GB3; англ. Globotriaosylsphingosine) в пятнах высушенной крови или плазме крови для биохимического подтверждения диагноза.
Также рекомендуется определение концентрации Lyso-GB3 в пятнах высушенной крови или плазме крови всем пациентам с установленным диагнозом болезни Фабри перед началом терапии и на фоне ФЗТ для биохимического контроля эффективности лечения. Количественное определение биомаркера Lyso-GB3 позволяет выявлять не только мужчин, но и женщин с болезнью Фабри, что важно при уточнении клинического статуса женщин-гетерозигот, а также для разрешения трудных диагностических случаев. Также Lyso-GB3 снижается в плазме крови на фоне адекватной ФЗТ и применяется в качестве показателя эффективности терапии и мониторинга состояния больных с болезнью Фабри.
Не рекомендуется проведение биопсии и патологоанат. исследования тканей почки и сердца для пациентов с клиническим подозрением на болезнь Фабри для подтверждения диагноза.
При наличии у пациента с болезнью Фабри признаков поражения почек с целью оценки состояния почек рекомендовано проведение лабораторных исследований согласно клиническим рекомендациям по ведению пациентов с ХПН не реже 1 раза в 6 мес.
Вне зависимости от клинических проявлений важно проводить регулярное определение количества белка и альбумина в суточной моче (микроальбуминурии/протеинурии), а также проведение оценки уровня расчетной СКФ исходно и далее не реже 1 раза в 6 мес всем пациентам с установленным диагнозом болезни Фабри и бессимптомным пациентам, выявленным при семейном скрининге для обнаружения патологии почек.
г) Инструментальные диагностические исследования. Для выявления поражения белого в-ва ГМ, ишемических очагов у пациентов с болезнью Фабри рекомендовано проводить МРТ ГМ при установлении диагноза и не реже 1 раза в год в случае, если изменения были выявлены до начала ФЗТ, или 1 раз в 36 мес, если изменения были выявлены после начала ФЗТ.
При МРТ ГМ у пациентов могут обнаруживаться ишемический инсульт, микрокровоизлияния и в/мозговые гематомы, изменения белого в-ва (лейкоареоз) и серого в-ва (кальцификация заднего таламуса), а также сосудистые мальформации, преимущественно представленные долихоэктазиями вертебробазилярной артерии. Эти изменения не являются высокосцецифичными для болезни Фабри, они могут встречаться у пациентов с др. патологией.
Рекомендована консультация врача-кардиолога не реже 1 раза в 6 мес и кардиологическое обследование, включающее ЭхоКГ (при первичном обследовании и далее не реже 1 раза в 12 мес), 12-канальную ЭКГ (при первичном обследовании и далее не реже 1 раза в 12 мес), холтеровское мониторирование (при первичном обследовании и далее не реже 1 раза в 12 мес) для своевременного выявления и/или мониторинга кардиологических изменений. В кардиологическое обследование также рекомендовано включать МРТ сердца (при первичном обследовании и далее не реже 1 раза в 12 мес) для своевременного выявления и/или мониторинга кардиологических изменений.
Всем пациентам с болезнью Фабри необходим осмотр врачом-офтальмологом (при первичном обследовании и далее не реже 1 раза в 3 года), включающий исследование переднего сегмента глаза методом бокового освещения, определение дефектов поверхности роговицы и обследование глазного дна (офтальмоскопию) с целью определения помутнения роговицы и др. глазной патологии. С помощью щелевой лампы выявляют помутнение роговицы в виде завитков. Сходные изменения роговицы наблюдаются при употреблении лекарств из группы хлорохинов или амиодарона.
Пациентам с болезнью Фабри в возрасте старше 5 лет рекомендована тональная аудиометрия при первичном осмотре и далее не реже 1 раза в 3 года с целью раннего выявления нарушений слуха. Детям с установленным диагнозом болезни Фабри рекомендуется консультация врача-педиатра, если ребенок не был осмотрен в предшествующие 6 мес для оценки общего состояния здоровья и определения тактики дальнейшей терапии.
В схему терапии болезни Фабри могут входить фенитоин и карбамазепин. Их применяют с целью снижения частоты приступов и выраженности хронической акропарестезии, а также периодических мучительных болевых кризов.
Пациентам с диспепсией назначают ЛП группы блокаторов Н2-гистаминовых рецепторов или ИПП. Для уменьшения протеинурии и лечения АГ применяют иАПФ и блокаторы рецепторов ангиотензина II, обладающие нефропротективными свойствами.
У пациентов с болезнью Фабри при фибрилляции предсердий рекомендуется проводить антикоагулянтную терапию варфарином.
д) Хирургическое лечение при болезни Фабри. Для пациентов с почечной недостаточностью жизненно важное значение имеют трансплантация почек и программный гемодиализ.
Пациентам с болезнью Фабри при развитии терминальной почечной недостаточности проводится трансплантация почек в соответствии с методическими рекомендациями «Трансплантация почки».
Пациентам с АВ-блокадой рекомендуется имплантация водителя ритма, а при наличии угрожающих жизни аритмий — кардиовертера-дефибриллятора в соответствии с разработанными клиническими рекомендациями для лечения данной патологии.
По желанию пациента удаляют ангиокератомы с помощью аргонового лазера.
У пациентов с болезнью Фабри можно применять ФЗТ ЛП рекомбинантной человеческой α-гал-А, продуцируемой в клетках яичников китайского хомячка [агалсидаза бета («Фабразим»), Джензим] и в клетках фибросаркомы человека [агалсидаза альфа («Реплагал»), «Шир ХГТ»]. Европейское агентство по лекарственным средствам одобрило к применению в странах Европейского союза и агалсидазу бета («Фабразим»), и агалсидазу альфа («Реплагал»), однако FDA одобрило к применению только агалсидазу бета («Фабразима»). Была продемонстрирована эффективность ФЗТ агалсидазой бета («Фабразимом») в отношении стабилизации заболеваний почек, регрессирования гипертрофической кардиомиопатии, уменьшения выраженности болевого синдрома и улучшения качества жизни.
ФЗТ проводят всем пациентам мужского пола после верификации диагноза лабораторными методами. У женщин ФЗТ начинают при наличии клинических проявлений заболевания, которые снижают качество жизни, и/или признаков прогрессирующего поражения органов-мишеней: кризов Фабри, и/или резистентной к стандартной терапии рецидивирующей или хронической невропатической боли в кистях и стопах, и/или персистирующей протеинурии, и/или снижения СКФ <80 мл/мин/1,73 м2, и/или поражения сердца, и/или нарушения мозгового кровообращения, и/или ишемических изменений ГМ, выявленных при МРТ.
ФЗТ не рекомендуется при беременности и в период кормления грудью; присутствии др. опасного для жизни заболевания, при котором прогноз вряд ли будет улучшен с помощью ФЗТ; у пациентов с серьезными осложнениями (напр., тяжелый инсульт, реанимационные пациенты).
Дозирование ЛП ФЗТ: агалсидазу альфа вводят в/в 0,2 мг/кг 1 р/2 нед, агалсидазу бета — в/в 1,0 мг/кг 1 р/2 нед. Первые введения ЛП должны быть сделаны в условиях стационара.
Оба ЛП не содержат консервантов и подлежат немедленному введению после приготовления р-ра для инфузии. В случае непереносимости возможен переход с одного ЛП на другой. Решение о замене ЛС принимается консилиумом специалистов, а пациент или его законные представители должны быть проинформированы о причине замены ЛП и дальнейшей тактике ведения.
Введение ФЗТ проводится регулярно при наличии показаний; в случае осложненного течения болезни — в условиях круглосуточного стационара, в стабильном состоянии — в стационаре дневного пребывания или амбулаторно 1 р/2 нед. Следует обращать внимание на соблюдение интервалов между инфузиями и недопустимость перерывов в терапии.
Все пациенты с болезнью Фабри должны проходить контрольные обследования (мониторинг) с целью оценки эффективности лечения. Переносимость ФЗТ обычно хорошая. Основными нежелательными эффектами могут быть реакции на введение ЛП (озноб, лихорадка, тошнота, тахикардия, зуд, миалгия, боли в конечностях, головная боль, боль в груди), в основном легкие или умеренно выраженные.
Поскольку большинство мужчин с классической формой заболевания не продуцируют ферментный белок, у этих пациентов в ответ на инфузии фермента могут синтезироваться IgG. Эти АТл, несмотря на очень высокие титры, не снижают эффективность клиренса субстрата. Лечение пациентов мужского пола с классическим фенотипом следует начинать в детстве.
е) Медицинская реабилитация, профилактика и диспансерное наблюдение. Медицинская реабилитация рекомендуется для пациентов с болезнью Фабри с выраженным болевым синдромом и склонностью к психоэмоциональным расстройствам. После установления диагноза пациенту с болезнью Фабри или его официальным представителям рекомендуется консультация врача-генетика с целью расчета генетического риска в семье, анализа родословной и обсуждения возможностей пренатальной диагностики.
ж) Примерный план обследования пациентов с болезнью Фабри. Первоначально до начала ФЗТ проводятся: подробный сбор анамнеза и оценка клинического статуса, оценка боли (визуально-аналоговая шкала оценки боли), определение активности фермента и ДНК-диагностика, микроальбуминурия/протеинурия, Расчетная СКФ, ЭКГ, ЭхоКГ, холтеровское мониторирование, МРТ сердца, МРТ ГМ, электронейромиография, осмотр окулиста, аудиограмма, ОАК и биохимический анализ крови.
Каждые 6 мес оцениваются следующие данные:
• сбор анамнеза и оценка клинического статуса (обязательно каждый визит);
• расчетная СКФ;
• ОАК и биохимический анализ крови;
• микроальбуминурия/протеинурия;
• ЭКГ чаще по показаниям.
Каждые 12 мес оцениваются следующие данные:
• оценка боли (визуально-аналоговая шкала оценки боли);
• ЭхоКГ чаще по показаниям;
• холтеровское мониторирование чаще по показаниям;
• МРТ сердца.
Каждые 36 мес оцениваются следующие данные:
• МРТ ГМ чаще по показаниям;
• осмотр окулиста чаще по показаниям;
• аудиограмма чаще по показаниям.