Термин миотубулярная миопатия является неправильным, потому что он подразумевает остановку созревания мышц плода во время миотубулярной стадии развития на 8-15-й неделе беременности. Этот термин основан на морфологическом облике миофибр в виде ряда центральных ядер и митохондрий внутри ядра цитоплазмы, с сократительными миофибриллами, образующими цилиндр вокруг этого ядра (рис. ниже). Эти морфологически аномальные миофибры не являются истинными фетальными миотрубками, поэтому более нейтральный и описательный термин центронуклеарная миопатия предпочтителен.
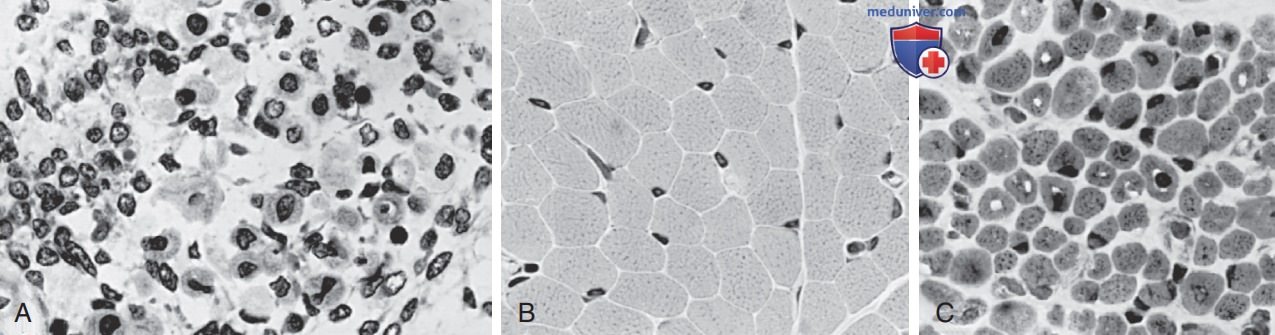
Поперечное сечение мышц: А — 14-нед плода человека; B — нормального доношенного новорожденного; C — доношенного новорожденного с Х-сцепленной рецессивной миотубулярной миопатией. Миофибры имеют крупные центральные ядра у плода и у больного миотубулярной миопатией, а ядра находятся на периферии мышечного волокна у новорожденного, как и у взрослого (гематоксилин и эозин, х500)
а) Патогенез. Общий патогенез включает потерю белка миотубуларина, что приводит к структурным и функциональным нарушениям в организации Т-канальцев и саркоплазматического ретикулума и дефектной связи возбуждения и сокращения. Хотя животные модели, выделенные для гена МТМ1, показывают значительное снижение высвобождения кальция, опосредованного рианодиновым рецептором 1, мутации в гене МТМ1 человека не влияют на гомеостаз и высвобождение кальция, опосредованное через рианодиновый рецептор 1, но они влияют на размер миотрубки и содержание ядер.
В истинных миотрубках плода периферическая миграция центральных ядер и ядра межъядерных митохондрий инициируется регрессией промежуточных филаментов фетального виментина на 15-20-й неделе беременности, которые удерживают эти структуры в центре миотрубки, но это не механизм центронуклеарных миопатий, за исключением, возможно, неонатальной миотонической дистрофии, которая включает в себя созревание некоторых миофибр.
б) Клинические проявления. Двигательная активность плода может уменьшаться на поздних сроках беременности. Многоводие является распространенным осложнением из-за слабости глотки плода и неспособности глотать околоплодные воды.
При рождении дети имеют тонкую мышечную массу, включающую осевые, поясные и дистальные мышцы конечностей; тяжелую генерализованную гипотонию и диффузную слабость. Дыхательные усилия м.б. неэффективными, требующими вентиляционной поддержки. Кормление через зонд может потребоваться из-за слабости мышц сосания и глотания. Яички часто не опущены. Лицевые мышцы м.б. слабыми, но у младенцев нет характерного лица миотонической дистрофии. Птоз м.б. заметной особенностью. Офтальмоплегия наблюдается в нескольких случаях. Нёбо м.б. высоким. Язык тонкий, но фасцикуляций не видно. Сухожильные рефлексы снижены или отсутствуют.
Миотубулярная миопатия не связана с кардиомиопатией (зрелые волокна сердечной мышцы обычно имеют центральные ядра), но в одном отчете описана полная АВ блокада без кардиомиопатии у пациента с подтвержденной Х-связанной миотубулярной миопатией. Врожденные аномалии ЦНС или др. систем с ней не связаны. Сообщалось об одном пациенте с прогрессирующей деменцией, у которого была мутация, удаляющая стартовый сигнал экзона 2. Известны пациенты с гораздо более мягкими симптомами или гораздо более поздним возрастом начала заболевания с мутациями в том же гене. Некоторые из них являются проявленными носителями.
в) Лабораторные результаты. Сывороточный уровень креатинкиназы в норме. ЭМГ не выявляет признаков денервации; результаты обычно нормальные или выявляют минимальные неспецифические миопатические признаки в раннем младенчестве. Скорость нервной проводимости м.б. медленной, но обычно нормальная. ЭКГ в норме. РОГК не обнаруживает кардиомегалии; ребра м.б. тонкими.
г) Диагноз. Если диагноз довольно вероятен по клинической картине, особенно если он подтвержден у родственника первой линии, генетические тесты м.б. проведены в неонатальном периоде. В большинстве случаев диагноз не так очевиден, но результаты биопсии мышц подтверждают диагноз при рождении, даже у недоношенных детей. Более 90% мышечных волокон мелкие и имеют центрально расположенные крупные везикулярные ядра в одном ряду. Пространства между ядрами заполнены саркоплазмой, содержащей митохондрии. Гистохимические пятна на окислительную ферментативную активность и гликоген обнаруживают центральное распределение, как и в миотрубках плода.
Цилиндр миофибрилл показывает зрелую гистохимическую дифференцировку с аденозинтрифосфатазными пятнами. Соединительная ткань мышечных веретен, кровеносных сосудов, в/мышечных нервов и моторных концевых пластин является зрелой. Ультраструктурные особенности, отличные от тех, которые определяют болезнь, также являются зрелыми. Электронная микроскопия показывает дезорганизованные триады и очаговую потерю миофиламентов. Виментин и десмин проявляют сильную иммунореактивность в мышечных волокнах при врожденной центронуклеарной миопатии и не проявляют заметной активности в мышцах новорожденных в нормальном сроке.
Несколько миотубуляринов присутствуют в циркулирующих тромбоцитах и могут оказаться простым неинвазивным скрининговым тестом у пациентов с подозрением на это заболевание. Молекулярно-генетический маркер в крови доступен также для ранней пренатальной диагностики, если имеется подозрение из-за семейного анамнеза. Пренатальная диагностика с помощью амниоцентеза возможна при явном подозрении на заболевание плода. Таблица 5 отличает центронуклеарную миопатию от др. врожденных миопатий.
д) Генетика. По меньшей мере 5 генов участвуют в патогенезе, что составляет ~80% пациентов. К ним относятся мутации в миотубуларине (ген МТМ1) с Х-сцепленными тяжелыми проявлениями; динамин 2 (DNM2) с АуД или спорадической встречаемостью; амфифизин 2 (BIN1) и титин (TTN) с АуР-наследственностью и рианодиновый рецептор 1 (RYR1) с АуР или спорадической встречаемостью.
Х-сцепленное рецессивное наследование является наиболее распространенной причиной заболевания, поражающего мужчин. Матери пострадавших младенцев клинически бессимптомны, но биоптаты их мышц показывают незначительные изменения. Генетическая связь на X-хромосоме была локализована в участке Xq28, локусе, отличном от гена Хр21 мышечных дистрофий Дюшенна и Беккера. Была идентифицирована делеция в ответственном гене МТМ1. Он кодирует белок под названием миотубуларин. Этот ген принадлежит к семейству аналогичных генов, кодирующих ферментативно активные и неактивные формы фосфатидилинозитол-3-фосфатаз, образующих димеры. МТМ1, динамин-2 и амфифизин локализуются на стенке Т-канальца в триадах.
Эта важнейшая область является местом, где потенциал действия высвобождает сигнал к рецептору рианодина для высвобождения кальция. Патогенез заключается в регуляции ферментативной активности и связывании с др. белками, индуцируемыми димерными взаимодействиями. Хотя задействован только 1 ген МТМ1, 5 различных точечных мутаций и множество разл. аллелей, а также большие дупликации могут привести к одному и тому же клиническому заболеванию. Мутации в белке динамин-2 приводят к АуД форме центронуклеарной миопатии и могут составлять 50% всех пациентов с центронуклеарной миопатией, но эти случаи обычно являются легкими и могут клинически не проявляться до взрослой жизни в виде диффузной, медленно прогрессирующей слабости и генерализованной мышечной псевдогипертрофии.
Известны и др. более редкие центронуклеарные миопатии; некоторые из них являются АуР и поражают оба пола, а др. спорадическими и неизвестного генетического происхождения. Рецессивные формы иногда делятся на раннюю форму с офтальмоплегией или без нее и позднюю форму без офтальмоплегии.
е) Лечение. В настоящее время доступно только поддерживающее и паллиативное лечение. Прогрессирующий сколиоз можно лечить задним спондилодезом. Генетические и нейропатологические исследования Х-сцепленной центронуклеарной (миотубулярной) миопатии привели к эффективной генной терапии у мышей и собак, так что животные в большей степени активны и у них в большей степени нарастает сила; терапия приводит к длительной экспрессии трансгена миотубуларина с нормальной мышечной работой и неврологической функцией при отсутствии мышечной патологии. Продолжаются испытания генной терапии Х-сцепленной центронуклеарной миопатии у людей.
ж) Прогноз. 75% тяжелобольных новорожденных с Х-сцепленной болезнью умирают в течение первых нескольких недель или месяцев жизни. У выживших не наблюдается прогрессирующего течения, но имеются серьезные физические недостатки, они редко ходят и остается выраженная гипотония. Поздние и особенно АуД формы имеют гораздо лучший прогноз, часто с легкой статической слабостью. Лечение генной терапией может кардинально изменить этот прогноз.