а) Гиперпитуитаризм. Первичная гиперсекреция гормонов гипофиза в педиатрической практике встречается редко, и ее следует отличать от вторичного гиперпитуитаризма. Он возникает вследствие физиологического ответа на дефицит гормонов периферических эндокринных желез. Этот дефицит приводит к нарушению обратной связи, например при гипогонадизме, гипокортицизме или гипотиреозе. При вторичном гиперпитуитаризме хроническая гиперсекреция возникает в ответ на дефицит гормонов периферических эндокринных желез.
Хроническая гиперсекреция приводит к гиперплазии гипофиза, вследствие чего может расширяться и разрушаться турецкое седло и в редких случаях повышаться ВЧД. Увеличение размеров не следует путать с первичными опухолями гипофиза. При назначении заместительной гормональной терапии и компенсации дефицита уровень гипофизарных гормонов легко снижается до нормальных значений и гиперплазия исчезает.
Первичная гиперсекреция гипофизарных гормонов аденомой относительно редко встречается в детстве. Наиболее часто диагностируемая в детстве аденома — пролактинома, за ней следует кортикотропинома, а затем соматотропинома. Они секретируют пролактин, кортикотропин и гормон роста соответственно. Имеется всего несколько сообщений о случаях тиреотропиномы у детей и подростков. Нет сообщений о гонадотропиномах у детей. Однако гамартомы гипоталамуса, избыточно секретирующие гонадотропин-рилизинг-гормон, являются одной из причин преждевременного полового созревания.
В очень редких случаях гиперплазия гипофиза может возникать в ответ на стимуляцию при эктопической продукции рилизинг-гормонов, которая иногда наблюдается у пациентов с синдромом Кушинга. Это состояние развивается вторично по отношению к избытку кортикотропин-рилизинг-гормона. Также вторично у детей с акромегалией оно развивается из-за избыточного образования соматотропин-рилизинг-гормона, который вырабатывается многими системными опухолями.
Моноклональная природа большинства аденом гипофиза предполагает, что большинство из них возникает в результате клонирования одной клетки. В некоторых случаях опухоли гипофиза возникают в результате стимуляции рилизинг-гормонами гипоталамуса, а в других, напр. при синдроме Мак-Кьюна-Олбрайта, образование опухоли обусловлено активирующей мутацией гена GNAS1, который кодирует α-субъединицу Gsα, гуанин-нуклеотид-связывающего белка. Клиническая картина обычно зависит от гипофизарного гормона, который чрезмерно секретируется.
Кроме того, в результате гормональной гиперсекреции или местного сдавливания опухолью часто возникают нарушения регуляции роста и/или полового развития. Синдром Мак-Кьюна-Олбрайта также характеризуется полиоссальной фиброзной дисплазией костей и пятнами цвета кофе с молоком с четким распределением.
б) Высокорослость. Согласно нормальному распределению, 2,3% населения будут на 2 SD (97,7%) выше среднего. Социальная приемлемость и даже желание быть выше (хайтизм) привели к тому, что жалобы на высокий рост не распространены в клинической практике. Обращаться за МП по поводу чрезмерного роста среди мужчин считается необычным. Исторически девушки (или их родители) чаще обращались к врачу с беспокойством по поводу высокого роста, но в наши дни даже среди них частота таких жалоб уменьшилась, поскольку высокий рост стал более приемлемым и социально желаемым для взрослых женщин.
Обеспокоенность по поводу побочных эффектов лечения эстрогенами и сообщения о неудовлетворенности взрослых женщин, получавших подобное лечение, привели к снижению использования эстрогенов для уменьшения конечного роста девочек, которые действительно чувствуют себя слишком высокими.
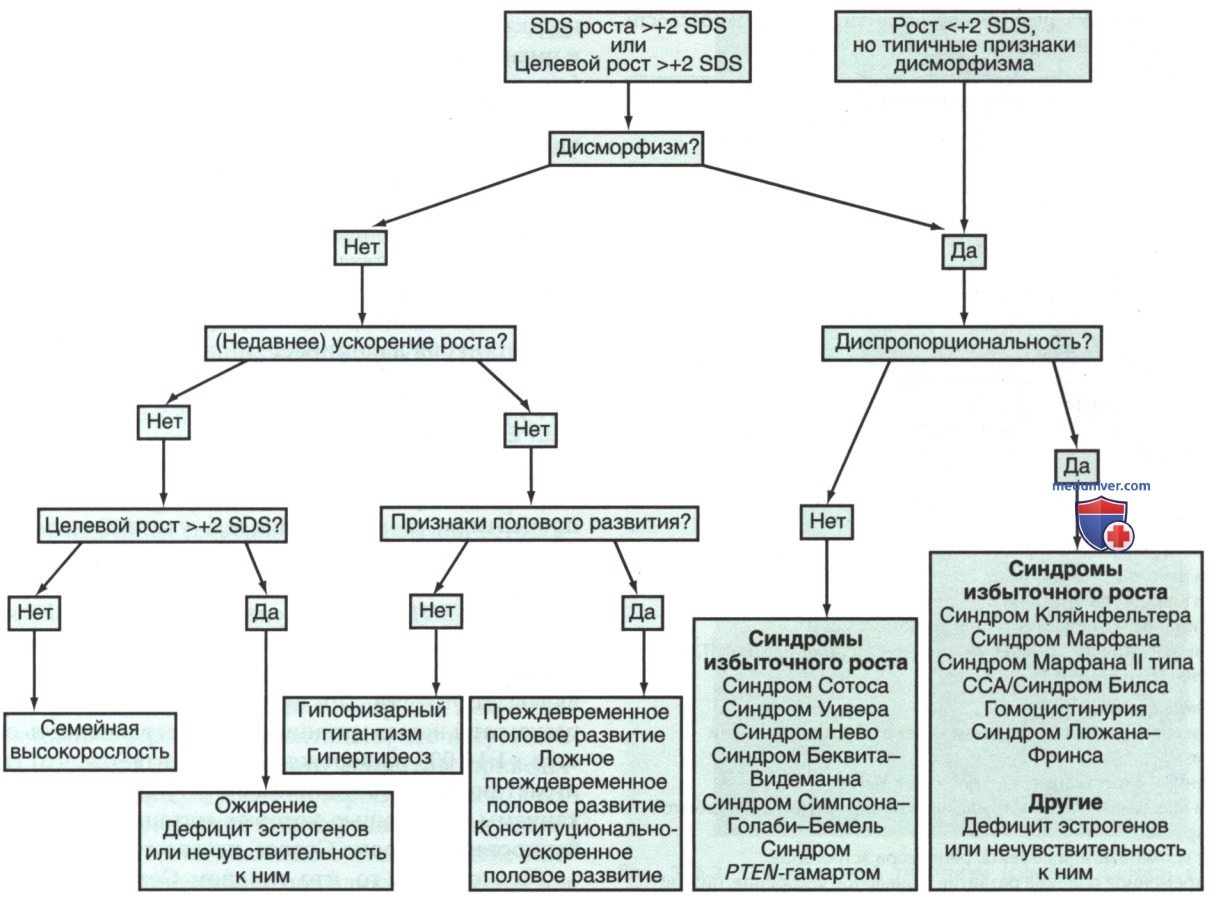
1. Дифференциальная диагностика высокорослости. В табл. 1 перечислены причины высокорослости при рождении, в детстве и подростковом возрасте. На рис. 1 показан алгоритм диагностики.
Рисунок 1. Алгоритм для дифференциальной диагностики высокорослости и синдромов избыточного роста. Целевой рост — процентиль настоящего роста >2 SDS от процентиля целевого роста, последний высчитывается на основе средне-родительского роста; SDS — коэффициент стандартного отклонения
- Чрезмерный рост плода и новорожденного. Диабет у матери является самой частой причиной рождения крупного для гестационного возраста ребенка. Даже при отсутствии клинических симптомов или семейного анамнеза рождение ребенка, крупного для гестационного срока, должно послужить причиной для обследования на наличие гестационного диабета.
- Синдромы избыточного роста: группа заболеваний, ассоциированных с чрезмерным соматическим ростом и ростом определенных органов, которые в совокупности называются синдромами избыточного роста. Эти заболевания чаще всего вызваны избыточной выработкой и доступностью инсулиноподобного фактора роста 2 (ИФР-2), кодируемого геном IGF2. Лучшим описанием этих синдромов является синдром Беквита-Видеманна, синдром избыточного роста с частотой встречаемости 1:13 700 новорожденных с равной вероятностью как для мальчиков, так и для девочек.
Он вызван генетическим или эпигенетическим изменением в хромосомном регионе 11р15; в большинстве случаев это связано с эпигенетическим дефектом (потеря или усиление метилирования ДНК) двух контролирующих областей импринтинга — IC1 и IC2. Другими причинами являются мутации, дупликация гена и потеря гетерозиготности в этом районе. Импринтированные гены, участвующие в развитии синдрома Беквита-Видеманна и ассоциированные с опухолями у детей, включают, помимо гена IGF2, ген Н19, который участвует в супрессии IGF2, а также WT-1 (ген опухоли Вильмса), ингибитор циклин-зависимой киназы 1С (CDKN1C), KQT-подобный член подсемейства калиевых потенциалзависимых каналов (KCNQ1) и KCNQ1-перекрывающий транскрипт 1 (KCNQ1OT1 или длинный интронный транскрипт QT, LIT1).
Около 15% случаев семейные, остальные спорадические.
Клинические признаки — проптоз с периорбитальной припухлостью (fullness), мальформация срединных глабеллярных капилляров (пламенеющий невус), складки и ямки мочки уха и макросомия, включая макроглоссию, гепатоспленомегалию, нефромегалию и омфалоцеле. У таких пациентов наблюдается вторичная гипогликемия вследствие гиперинсулинемии, развивающейся в результате гиперплазии β-клеток ПЖЖ.
Дети отличаются предрасположенностью к росту эмбриональных опухолей, включая опухоль Вильмса, гепатобластому и адренокортикальную карциному. Основное внимание уделяется омфалоцеле, проблемам с ДП (в результате макроглоссии) и неонатальной гипогликемии.
Риск развития рака до возраста 8 лет высокий, поэтому до достижения этого возраста рекомендуется регулярное наблюдение, которое включает УЗИ БП и измерение а-фетопротеина каждые 3 мес. В дальнейшем каждые 1-2 года рекомендуется проводить УЗИ почек, т.к. позже могут развиться медуллярные губчатые почки и нефрокальциноз.
Мутации в гене глипикана GPC3 (который кодирует ИФР-2-нейтрализующий мембранный рецептор), вызывает синдром избыточного роста Симпсона-Голаби-Бемель. Другие синдромальные причины избыточного роста плода включают синдромы Костелло, Уивера, Сотоса и Перлмана.
- Избыточный рост в детстве и в подростковом возрасте. Вариант нормы, семейная или конституциональная высокорослость является наиболее частой причиной высокого роста. Почти всегда в семейном анамнезе есть родственники высокого роста, и отсутствует органическая патология. В детстве ребенок часто выше сверстников и обладает хорошим здоровьем. Физикальное и лабораторное обследования (если они проводятся) не выявляют отклонений от нормы.
- Экзогенное ожирение ассоциировано с быстрым линейным ростом и относительно ранним началом полового развития (чаще у девочек). Наблюдается ускорение костного возраста, что приводит к высокому росту в детстве, однако конечный рост обычно нормальный.
- Синдром Кляйнфельтера (синдром XXY) — относительно распространенная (1—2:1000 живорожденных мальчиков) хромосомная патология, ассоциированная с высоким ростом, трудностями обучения (включая необходимость помощи логопеда), гинекомастией и сниженным соотношением верхнего сегмента к нижнему. У мальчиков может наблюдаться гипотония, клинодактилия и гипертелоризм. Яички всегда маленькие, хотя выработка андрогенов клетками Лейдига часто находится в низко-нормальном диапазоне. Сперматогенез и функция клеток Сертоли нарушены и приводят к бесплодию. Другие нарушения полового развития включают относительно небольшой размер полового члена и высокую частоту гипоспадии и крипторхизма.
- Синдром XYY ассоциирован с высоким ростом, выраженным акне в подростковом возрасте, проблемами в обучении, поведении, особенно связанными с импульсивностью. Интеллект обычно средний, однако м.б. ниже на 10-15 баллов IQ, чем у их братьев и сестер. Другие редкие хромосомные патологии, при которых встречаются лишние X- или У-хромосомы (напр., XXX, XXXY, XYYY), также ассоциируются с высоким ростом.
- Синдром Марфана — АуД-заболевание соединительной ткани, при котором наблюдается высокий рост, арахнодактилия, тонкие конечности, увеличенный размах рук и уменьшение соотношения верхнего сегмента к нижнему. Дополнительные нарушения включают аномалии глаз (напр., подвывих хрусталика), гипотонию, кифосколиоз, деформации клапанов сердца и дилатацию корня аорты.
- Гомоцистинурия — АуР врожденное нарушение метаболизма аминокислот, вызванное дефицитом фермента цистатион β-синтетазы. При отсутствии лечения характерна умственная отсталость. По многим клиническим признакам, особенно по аномалиям глаз, гомоцистинурия напоминает синдром Марфана.
в) Синдром Сотоса (церебральный гигантизм). У детей с церебральным гигантизмом (также известным как синдром Сотоса) вес и рост при рождении >90-го процентиля, размер головы м.б. увеличенным. В других случаях макрокрания становится более заметной позже. Большинство случаев синдрома Сотоса вызвано мутациями гена NSD1 (белок 1, содержащий домен SET ядерного рецептора), однако в японской популяции большинство случаев связано с микроделециями области 5q35, которая включает этот ген. Тип наследования АуД, 95% случаев — результаты новых мутаций. Распространенность оценивается 1:14 000 живорожденных. Считается, что ген NSD1 играет роль в эпигенетической регуляции, но точные механизмы, с помощью которых мутации приводят к особенностям синдрома Сотоса, неизвестны.
Несмотря на то, что синдром Сотоса характеризуется быстрым ростом, никаких доказательств эндокринной дисрегуляции при этом синдроме нет. Рост заметно ускорен. К 1 году жизни рост младенцев с данным синдромом становится >97-го процентиля. Ускоренный рост продолжается первые 4-5 лет, а затем возвращается к нормальным темпам (рис. 2). Половое развитие начинается в обычные сроки, но может начаться немного раньше. Конечный рост ближе к верхней границе нормы.
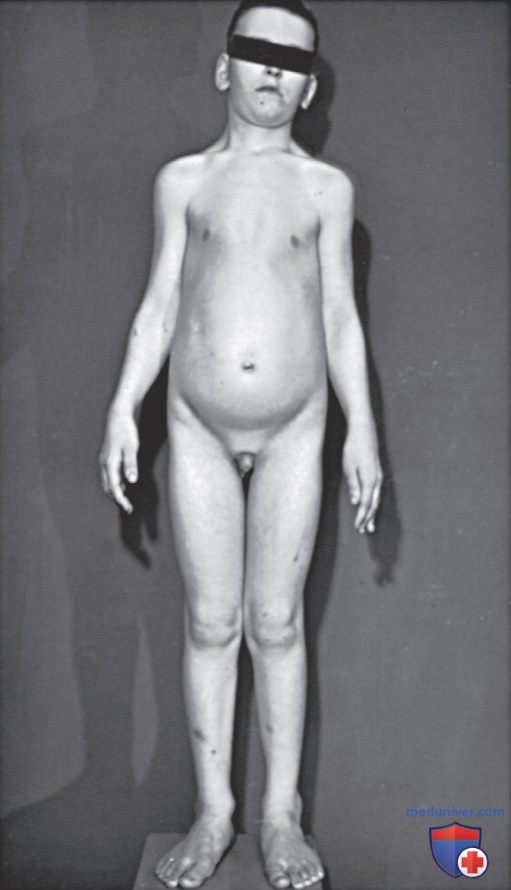
Рисунок 2. Церебральный гигантизм (синдром Сотоса) у мальчика 8 лет. Рост и костный возраст соответствовали 12 годам. Уровень IQ = 60. По данным электроэнцефалографии выявлены нарушения. Обратите внимание на выступающие лоб и челюсть, а также на большие руки и ноги. Половое развитие соответствовало хронологическому возрасту. При исследовании гормонального профиля все показатели были в норме. Конечный рост 208 см (6 футов 10 дюймов); половое развитие было нормальным. Размер ноги — 18
Клинически синдром характеризуется большой (макроцефалия) долихоцефальной головой, выступающим лбом и челюстью, гипертелоризмом, антимонголоидным разрезом глаз, готическим нёбом, большими кистями и стопами с утолщенной ПЖК. Также отмечаются неповоротливость и неуклюжая походка. Дети с этим синдромом испытывают большие трудности при занятиях спортом, с обучением езде на велосипеде и др. задачами, требующими координации. У большинства пациентов имеется некоторая умственная отсталость; у части детей могут преобладать нарушения восприятия. У пациентов было зарегистрировано множество различных типов нефебрильных судорог. До 25% пациентов с синдромом Сотоса имеют судорожные приступы в течение жизни.
У пациентов с этим синдромом риск развития новообразований, включая нейробластомы, гепатобластомы и лейкемию, повышен и составляет 2-4%. Созревание костной ткани обычно соответствует росту пациента, хотя имеются сообщения об ускорении костного возраста. Сколиоз развивается в 30% случаев, обычно начиная со школьного возраста. Уровень гормона роста, инсулиноподобного фактора роста 1 и др. гормонов обычно в пределах нормы; отсутствуют четкие лабораторные или рентгенологические маркеры данного синдрома. Часто встречаются изменения ЭЭГ; при визуализации ГМ нередко обнаруживают увеличение желудочковой системы, однако при этом ВЧД нормальное.
Доступно и широко используется генетическое исследование для обнаружения мутаций в гене NSD1 (или флуоресцентная гибридизация in situ для поиска микроделеций 5q35 у пациентов из Японии).
Лечение симптоматическое и включает повышенное внимание к проблемам развития и поведения (которые с возрастом улучшаются), сколиозу и эпилепсии. Высокорослость не требует специфического лечения. В настоящее время не существует единого мнения о необходимости скрининга на новообразования.
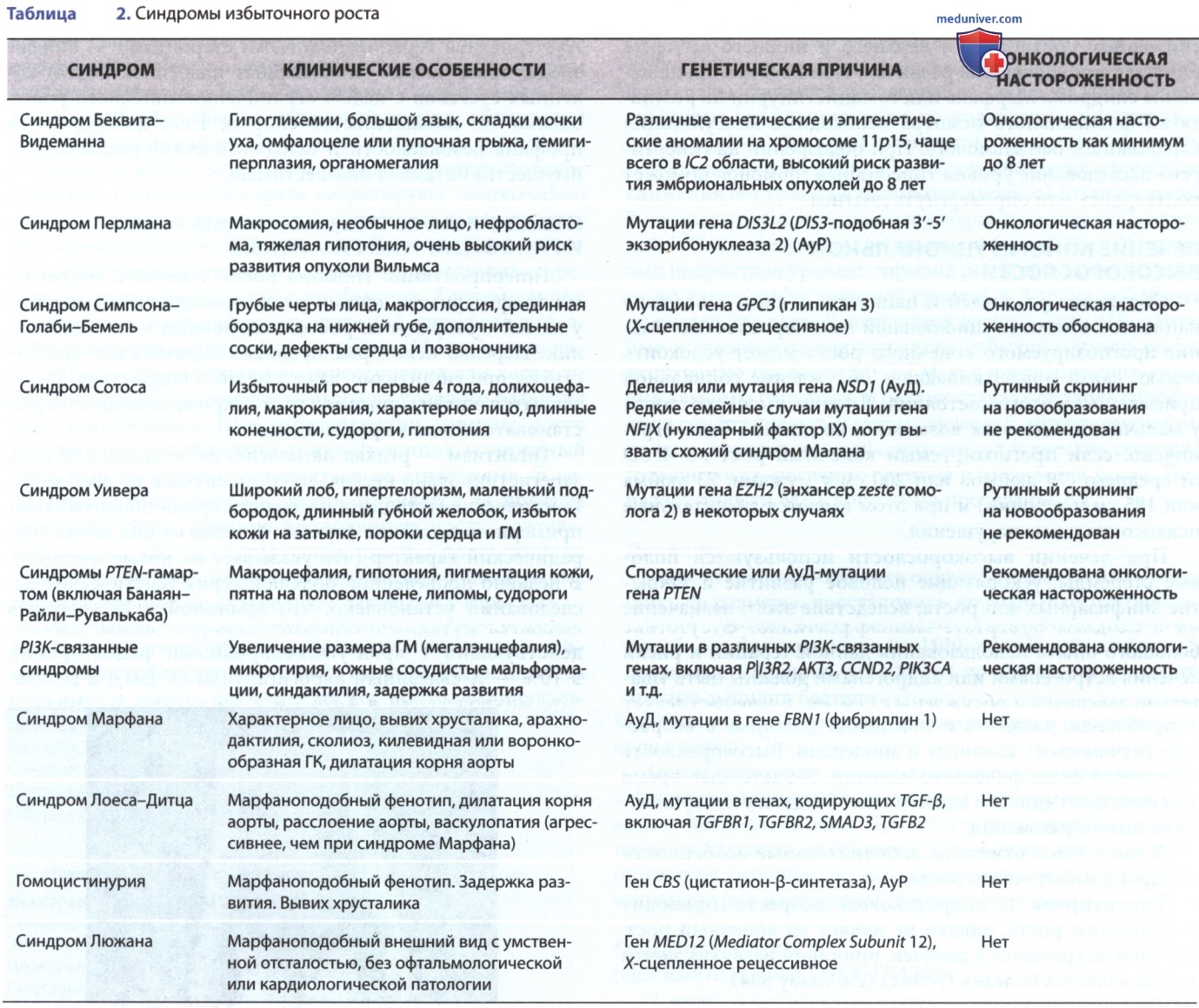
В табл. 2 отмечены дополнительные особенности синдрома избыточного роста.
Гипертиреоз в подростковом возрасте приводит к ускорению роста, однако не влияет на конечный рост. Он чаще встречается у девочек, причиной развития почти всегда является болезнь Грейвса.
При преждевременном половом развитии, центральном (повышенная секреция гонадотропинов) или периферическом (повышенная секреция андрогенов или эстрогенов, или того и другого), наблюдается ускорение линейного роста в детстве, имитирующее пубертатный скачок роста. Ускорение костного возраста отрицательно влияет на конечный рост.
Несмотря на то, что при задержке полового развития, как и при конституциональной задержке, может наблюдаться низкий рост, есть вероятность, что отсутствие пубертата и завершения полового развития приведет к продолжению процессов роста во взрослой жизни, а соответственно, и к выраженной высокорослости. У представителей как мужского, так и женского пола закрытие эпифизарных зон роста происходит под влиянием эстрогенов (образующихся из тестостерона и других андрогенов путем ароматизации), поэтому редко встречаемые дефекты ферментов ароматазы или рецепторов эстрогенов могут привести к нарушению этого процесса и высокому росту, при этом вследствие отсутствия закрытия ростовых зон высокие темпы роста сохраняются и во взрослом возрасте.
- Диагностическое обследование: целью обследования является ДД часто встречающейся конституциональной нормы и редких патологических состояний. Зачастую, если анамнез свидетельствует о семейной высокорослости, а физикальное обследование не выявляет патологии, нет необходимости проводить лабораторные исследования. Важно провести рентгенографическое исследование для оценки костного возраста, определение прогнозируемого конечного роста, который служит основой тактики ведения при обсуждении данного вопроса с родителями. Если в анамнезе имеются признаки какого-либо из перечисленных расстройств или при физикальном осмотре выявляют отклонения, должны быть проведены лабораторные исследования.
Скрининговым методом выявления избытка гормона роста является определение уровней инсулиноподобного фактора роста 1 и ИФР-связывающего белка 3; верифицировать данное состояние можно проведением супрессивного теста с декстрозой («Глюкозой»). При лабораторном подтверждении избыточной секреции гормона роста требуется проведение МРТ для оценки гипофиза. Для исключения синдрома Кляйнфельтера у мальчиков следует провести хромосомный анализ, особенно при сниженном соотношении верхнего и нижнего сегмента или наличии нарушения развития. При подозрении на наличие синдрома Марфана или гомоцистинурии по результатам физикального осмотра необходима консультация кардиолога и офтальмолога. При подозрении на гипертиреоз исследование уровня тиреоидных гормонов поможет подтвердить или опровергнуть диагноз.
г) Лечение конституционной высокорослости. Ободрение родителей и пациента — ключ к ведению пациентов с конституциональной высокорослостью. Знание прогнозируемого конечного роста может успокоить семью, как и поддерживающее обсуждение социальной приемлемости этого состояния. Лечение высокорослости у мальчиков и девочек возможно, но должно быть ограничено, если прогнозируемый конечный рост >3-4 SD от среднего (79 дюймов или 200 см у мужчин, 73 дюйма или 185 см у женщин) и при этом имеются значительные психосоциальные нарушения.
При лечении высокорослости используются половые стероиды, ускоряющие половое развитие и закрытие эпифизарных зон роста; вследствие этого назначение их в позднем пубертате малоэффективно. Отсутствие большого опыта использования данной терапии и риски лечения эстрогенами или андрогенами должны быть тщательно взвешены и обсуждены с семьей, а лечение следует рекомендовать только в исключительных случаях. Также желательно детальное обсуждение с ребенком на его уровне, так как до 40% пациентов, получавших лечение, во взрослом возрасте недовольны и считают, что были недостаточно проконсультированы по поводу такой тактики ведения. По возможности терапию начинают до начала полового развития или в раннем пубертате (при костном возрасте не старше 14 лет).
В крайне редких случаях у мальчиков по желанию используется тестостерона энантат в дозе 250-500 мг в/м 1 р / 2 нед на протяжении 6 мес. У девочек с целью уменьшения прогнозируемого роста используется пероральный прием разных доз эстрогенов, однако конечный рост снижается всего на 1,1-2,4 см. Терапию необходимо начинать при костном возрасте <12 лет. В редких случаях, когда рекомендована терапия, используется пероральный прием этинилэстрадиола в дозе 0,15-0,5 мг/сут до закрытия зон роста. К кратковременным побочным эффектам относятся доброкачественные заболевания груди, ЖКБ, АГ, нарушения менструального цикла, набор веса, тошнота, боль в конечностях, галакторея и тромбоз. Потенциальным долгосрочным осложнением является снижение фертильности во взрослом возрасте.
Альтернатива терапии половыми стероидами — эпифизиодез (разрушение эпифизарных пластинок роста) коленных суставов с целью ограничения линейного роста, однако это вмешательство спорно, а его долгосрочный профиль безопасности и психологические риски и преимущества остаются неизвестными.
д) Избыточная секреция гормона роста и гипофизарный гигантизм. Гиперпродукция гормона роста у детей с открытыми эпифизарными зонами роста приводит к гигантизму; у людей с закрытыми ростовыми зонами — к акромегалии. Нередко некоторые признаки акромегалии присутствуют при гигантизме, даже у детей и подростков. После закрытия эпифизарных зон роста акромегалоидные черты становятся более выраженными.
Гигантизм — редкая патология, на сегодняшний день зарегистрировано несколько сотен случаев во всем мире. Генетические мутации в настоящее время обнаруживают примерно в половине случаев. Многие из них носят спорадический характер (что указывает на новые мутации). В недавно проведенной большой серии генетических исследований установлено, что причиной в 29% случаев являются мутации в гене AIP (кодирует белок, взаимодействующий с арил-углеводородными рецепторами), в 10% — Х-связанный акрогигантизм (X-LAG) в результате микроделеций в Хq26.3 и в 5% случаев — синдром Мак-Кьюна-Олбрайта. В 54% случаев при проведении генетического исследования мутаций не выявлено.
Хотя с течением времени аденомы, секретирующие гормон роста, развиваются почти у 60% пациентов с синдромом множественной эндокринной неоплазии 1-го типа, большинство из них возникают во взрослом возрасте и поэтому вызывают акромегалию, а не гигантизм.
Повышенная секреция гормона роста и секретирующие гормон роста аденомы также могут наблюдаться при нейрофиброматозе, туберкулезном склерозе и Карни-комплексе.
- Клинические проявления: основное клиническое проявление гигантизма — ускорение линейного роста вследствие избытка гормона роста. Типичными проявлениями являются грубые черты лица и увеличение размеров кистей и стоп. У маленьких детей ускорению линейного роста может предшествовать увеличение размера головы. У некоторых пациентов наблюдаются поведенческие проблемы и нарушения зрения. В большинстве зарегистрированных случаев аномальный рост выявлялся в период полового развития, однако одному ребенку данный диагноз был установлен в период новорожденности, а другому в возрасте 21 мес.
Есть несколько сообщений о том, что пациенты с данной патологией достигали высоты более 8 футов (243 см). В некоторых случаях основными жалобами пациента м.б. симптомы, характерные для местных проявлений опухоли гипофиза (головная боль, нарушение полей зрения и дефициты др. тропных гормонов гипофиза). Было зарегистрировано одно сообщение о пациенте с диабетическим кетоацидозом, вызванным избытком гормона роста. Проявления гигантизма обычно привлекают больше внимания, чем постепенно развивающаяся акромегалия у взрослых.
Секретирующие гормон роста аденомы гипофиза у мальчиков встречаются чаще, однако у девочек они могут возникать в более раннем возрасте. Опухоли при мутации гена AIP чаще встречаются у мальчиков, они больше и инвазивнее секретируют гормон роста или пролактин. X-LAG-синдром бы недавно признан причиной семейных аденом гипофиза, причем он чаще встречается у пациентов женского пола и характеризуется быстрым ростом в младенческом возрасте. У пациентов с синдромом Мак-Кьюна-Олбрайта обычно наблюдаются и др. проявления синдрома, такие как полиоссальная фиброзная дисплазия, пятна цвета кофе с молоком и преждевременное половое развитие. Опухоли гипофиза секретируют чрезвычайно высокие уровни гормона роста (сообщалось об уровнях до 1500 мкг/л), и ~в 50% при аденомах гипофиза наблюдается гиперпролактинемия, поскольку происходит секреция как гормона роста, так и пролактина.
Аденомы могут приводить к нарушению др. функций гипофиза вследствие роста или кистозной дегенерации. Может нарушаться секреция гонадотропинов, тиреотропина или кортикотропина. Может развиваться задержка полового развития или гипогонадизм. При гиперсекреции гормона роста в сочетании с дефицитом гонадотропинов ускоренный линейный рост может сохраняться десятилетиями. В некоторых случаях опухоль распространяется за пределы турецкого седла, поражая клиновидную кость, зрительные нервы и ГМ. Секретирующие гормона роста опухоли у детей с большей вероятностью локально инвазивные или агрессивные, по сравнению с таковыми у взрослых.
Признаком акромегалии в основном является увеличение дистальных частей тела, однако проявления аномального роста наблюдаются во всех отделах. Увеличивается окружность черепа, расширяется нос, часто увеличивается язык, черты лица становятся грубее. Нижняя челюсть чрезмерно увеличивается в размерах, происходит расширение промежутков между зубами. Часто встречаются дефекты полей зрения и неврологические нарушения; признаки повышенного ВЧД появляются позднее. Пальцы рук и ног в основном утолщаются. Может быть дорсальный кифоз. Ранние симптомы — усталость и утомление. Уровни гормона роста повышены иногда >100 нг/ мл. Обычно не наблюдается подавления уровня гормона роста в ответ на гипергликемию или глюкозотолерантный тест, а уровни инсулиноподобного фактора роста 1 и ИФР-связывающего белка 3 при акромегалии и гипофизарном гигантизме постоянно высокие.
1. Диагностика. Большинство детей с высоким ростом не имеют гипофизарного гигантизма, поэтому следует исключать др. причины быстрого линейного роста, такие как генетические причины высокорослости, преждевременное половое развитие и гипертиреоз. Сопутствующие данные (напр., дисморфические черты лица, нейрокогнитивные расстройства, гемигипертрофия) могут указывать на синдромальные или хромосомные причины высокого роста, такие как синдром Сотоса, Уивера, Кляйнфельтера или синдром XYY. Наличие гиперсекреции гормона роста можно исследовать путем определения уровней инсулиноподобного фактора роста 1 и ИФР-связывающего белка 3. Повышенный уровень инсулиноподобного фактора роста 1 у пациента с подозрением на наличие заболевания и соответствующей клинической картиной обычно указывает на избыток гормона роста.
Возможная путаница может возникнуть при оценке обычных подростков, поскольку в период пубертата наблюдаются более высокие, в сравнении со взрослыми людьми, уровни инсулиноподобного фактора роста 1, поэтому его уровни следует соотносить с возрастом и полом. Уровень ИФР-связывающего белка 3 в сыворотке также является чувствительным маркером повышения гормона роста, он будет повышен почти во всех случаях. Если уровни инсулиноподобного фактора роста 1 и/или ИФР-связывающего белка 3 повышены, то следующим шагом является исследование секреции гормона роста путем проведения теста на подавления с декстрозой («Глюкозой»). «Золотым стандартом» диагностики гиперсекреции гормона роста у взрослых считается отсутствие снижения его уровня в сыворотке крови <1 нг/дл во время 2-часового орального глюкозотолерантного теста с нагрузкой 1,75 г/кг декстрозы («Глюкозы») (максД 75г).
У здоровых подростков уровень гормона роста может не снизиться до таких цифр, поэтому уровень в 5 нг/мл м.б. более приемлем для данной возрастной группы. Если лабораторные исследования подтверждают избыток гормона роста, необходимо провести МРТ ГМ для подтверждения наличия аденомы гипофиза. В редких случаях опухоль гипофиза не обнаруживают. В таком случае м.б. микроаденома гипофиза, которая не видна, или же эктопическая секреция гормон роста-релизинг-гормона или гормона роста. При недоступности МРТ допустимо проведение КТ.
2. Лечение. Цели терапии — удалить опухоль гипофиза или уменьшить ее размеры, восстановить нормальный паттерн секреции гормона роста, нормализовать уровни инсулиноподобного фактора роста 1 и ИФР-связывающего белка 3, сохранить секрецию гипофизом др. гормонов и предотвратить рецидив болезни.
Если аденомы гипофиза имеют четкие контуры, то методом выбора становится трассфеноидальная хирургия, которая может привести к полному излечению. Опухоль следует удалить полностью. Эффективность хирургического лечения во многом зависит от опытности хирурга, равно как и от размера и степени распространения опухоли. Интраоперационное измерение уровня гормона роста может улучшить исход резекции опухоли. Транссфеноидальная операция по удалению опухоли так же безопасна у детей, как и у взрослых. В некоторых случаях может потребоваться транскраниальный доступ. Основная задача лечения — нормализация уровней гормона роста и инсулиноподобного фактора роста 1.
Лучшими биохимическими показателями для определения эффективности лечения являются уровни гормона роста [<1 нг/мл через 2 ч после нагрузки декстрозой («Глюкозой»)] и инсулиноподобного фактора роста 1 (соответствие возрастной норме).
Если после операции не происходит нормализация уровней гормона роста и инсулиноподобного фактора роста 1, возможно облучение гипофиза и медикаментозная терапия. Лучевая терапия предотвращает дальнейший рост опухоли в >99% случаев. Основной недостаток — медленное снижение уровня гормона роста на 50% от исходного через 2 года, на 75% через 5 лет и 90% через 15 лет. Через 10 лет после облучения у 40-50% пациентов предсказуемо возникает множественный дефицит гормонов гипофиза.
Хирургическое лечение неэффективно в значительном числе случаев, а ответ на лучевую терапию может появляться недостаточно быстро, поэтому медикаментозная терапия играет важную роль в лечении пациентов с избыточной секрецией гормона роста. Лечение антагонистами гормона роста, аналогами соматостатина длительного действия и, в некоторых случаях, агонистами дофамина эффективно и хорошо переносится.
Пэгвисомант является антагонистом рецептора гормона роста, который конкурирует с эндогенным гормоном роста за связывание с его рецептором. Он эффективно подавляет уровни гормона роста и инсулиноподобного фактора роста 1 у пациентов с акромегалией, вызванной опухолями гипофиза, а также эктопической гиперсекрецией гормон роста-релизинг-гормона. Нормализация уровня инсулиноподобного фактора роста 1 происходит у 90% пациентов, ежедневно принимающих этот препарат >3 мес. Дозировка для взрослых 10-40 мг п/к 1 p/день, хотя протоколы, в которых инъекции проводили 2 р/нед, также были очень успешны.
Необходимо контролировать уровни инсулиноподобного фактора роста 1 и печеночных ферментов. Также эффективна комбинированная терапия аналогами соматостатина и еженедельными инъекциями пэгвисоманта.
Опыт применения в педиатрии ограничен, однако описания клинических случаев показывают, что он может успешно подавлять уровни инсулиноподобного фактора роста 1 при использовании в дозах 10-30 мг/сут.
Аналоги соматостатина часто эффективны при лечении пациентов с избыточной продукцией гормона роста. Октреотид подавляет секрецию гормона роста <2,5 нг/мл у 65% пациентов с акромегалией и нормализует уровень инсулиноподобного фактора роста 1 у 70%. Эффект октреотида сохраняется с течением времени. При применении октреотида также происходит уменьшение размеров опухоли, но оно обычно незначительное. Постоянное подавление секреции гормона роста м.б. достигнуто путем непрерывной п/к-инфузии октреотида с помощью помпы или при использовании препаратов пролонгированного действия, включая октреотид длительного действия и ланреотид.
Инъекции октреотида у детей применялись в дозах 1-40 мкг/кг/сут. Препараты пролонгированного действия для взрослых используются в дозе 10-40 мг ежемесячно; диапазон доз для детей не установлен.
В отношении пациентов с одновременной гиперсекрецией гормона роста и пролактина можно использовать агонисты дофамина, напр. бромокриптин и каберголин, которые связываются с рецепторами дофамина 2-го типа гипофиза и также могут подавлять секрецию гормона роста. Часто происходит адекватное подавление уровней пролактина, но при использовании только этого лечения уровни гормона роста и инсулиноподобного фактора роста-1 редко нормализуются. Уменьшение размеров опухоли происходит у меньшей части пациентов. Эффективность этих препаратов можно сочетать с эффективностью октреотида. Терапия каберголином в дозах 0,254,0 мг/нед (1-2 р/нед) использовалась у взрослых с акромегалией, и вследствие менее частого приема и более низкой частоты побочных эффектов по сравнению с бромокриптином этот агонист дофамина считается препаратом выбора как у взрослых, так и у детей. Побочные эффекты могут включать тошноту, рвоту, боль в животе, аритмии, заложенность носа, ортостатическую гипотензию, нарушения сна и утомляемость.
е) Гиперсекреция других гормонов гипофиза:
1. Пролактинома. Аденомы гипофиза, секретирующие пролактин, — самые частые опухоли гипофиза у подростков. С появлением МРТ выявляется больше таких опухолей, особенно микроаденом (<1 см в диаметре). Наиболее частыми проявлениями являются головная боль, первичная или вторичная аменорея и галакторея. У девочек данное заболевание встречается в >2 раза чаще, чем у мальчиков; при этом у большинства пациентов симптомы появляются уже после периода полового созревания. Лишь у немногих наблюдается задержка полового развития. У некоторых семей с синдромом множественной эндокринной неоплазии I типа пролактиномы проявляются в подростковом возрасте. Семейные случаи, а также спорадические случаи мутаций de novo гена AIP и X-LAG выявляются все чаще, поскольку генетическое тестирование становится более распространенным.
Уровень пролактина м.б. повышен умеренно (40-50 нг/мл) или значительно (10 000-15 000 нг/мл), а также существует корреляция между размером опухоли и уровнем пролактина. Большинство пролактином у детей имеют большие размеры (макроаденомы), вызывают увеличение турецкого седла, а в некоторых случаях — дефекты полей зрения. Примерно у 30% пациентов с макроаденомами развивается дефицит других гормонов гипофиза, в частности дефицит гормона роста. С другой стороны, пролактинсекретирующие аденомы могут также избыточно секретировать гормон роста и/или ТТГ.
Пролактиномы следует отличать от гиперпролактинемии и гиперплазии гипофиза, которые могут возникнуть у пациентов с первичным гипотиреозом, который лечится тиреоидными гормонами. Умеренное повышение пролактина (<200 нг/мл) также связано с приемом различных ЛП (нейролептики, метоклопрамид, фенотиазины, верапамил), с нарушением целостности воронки гипофиза, напр. при краниофарингиоме, с хроническим стрессом (редко >40 нг/мл), стимуляцией сосков, и в ряде случаев м.б. идиопатическим.
В некоторых случаях выраженная гиперпролактинемия ассоциирована с hook-эффектом, который приводит к ложнонизким показателям в анализах крови. В случаях, когда клинические признаки соответствуют гиперпролактинемии, следует выполнить последовательное раз-ведение лабораторного образца для исключения ошибки измерения. С другой стороны, пациенты могут иметь ложноповышенные уровни пролактина при проведении иммуноанализа вследствие наличия полимеров и димеров пролактина (макропролактинемия), которые не являются физиологически активными. При обнаружении повышенного уровня пролактина у пациента без симптомов можно избежать ненужных диагностических обследований и лечения, выполнив осаждение полиэтиленгликолем с целью исключить наличие макропролактинемии, которая является клинически доброкачественной.
У большинства пациентов, у которых гиперпролактинемия является вторичной вследствие аденомы, возможно эффективное лечение агонистами дофамина. Лечение приводит к снижению уровня пролактина и уменьшению размеров опухоли у подавляющего большинства пациентов. Благодаря большей эффективности и меньшей частоте побочных эффектов каберголин считается препаратом выбора для лечения гиперпролактинемии. Общепринятый протокол — начинать с 0,25 мг 2 р/нед, а затем увеличивать дозу до 1 мг 2 р/нед. Некоторым пациентам могут потребоваться более высокие дозы, но их следует тщательно контролировать; высокие дозы, применяемые длительно у пожилых пациентов с паркинсонизмом, ассоциированы с поражением сердечных клапанов, хотя нет сообщений об этом при применении доз, используемых для лечения гиперпролактинемии у детей; мониторинг состояния сердечных клапанов с помощью ЭхоКГ м.б. целесообразен при длительном назначении высоких доз.
Если лечение агонистами дофамина не приводит к снижению концентрации пролактина в сыворотке или уменьшению размера аденомы и когда симптомы или признаки, характерные для гиперпролактинемии или размера аденомы, сохраняются во время лечения, можно рассмотреть возможность проведения трансфеноидальной хирургической операции. В очень редких случаях злокачественной пролактиномы может потребоваться XT темозоломидом, но добиться выздоровления в таких случаях сложно.
2. Кортикотропинома. Кортикотропиномы очень редко встречаются у детей, пик возникновения приходится на возраст 14 лет. К болезни Кушинга приводит, в частности, аденома гипофиза, секретирующая АКТГ, который стимулирует избыточную продукцию и выработку кортизола. Аденомы встречаются чаще, чем первичные надпочечниковые причины синдрома Кушинга, за исключением детей младшего возраста (<5 лет), у которых карциномы надпочечников и активирующие мутации при синдроме Мак-Кьюна-Олбрайта являются редкими, но доминирующими причинами синдрома. Аденомы, вызывающие болезнь Кушинга, почти всегда представляют собой микроаденомы диаметром <5 мм, на момент появления клинических проявлений их размер значительно меньше, чем при других типах аденом. Наиболее чувствительным индикатором избыточной секреции ГКС у детей является задержка роста, которая обычно предшествует другим проявлениям. У пациентов наблюдается набор веса, причем скорее центрального типа, чем генерализованного.
Также часто наблюдаются задержка полового развития, гипертония, широкие багровые стрии, утомляемость и депрессия. Среди детей препубертатного возраста мальчики болеют чаще, чем девочки.
Определение кортизола в слюне в полночь можно использовать в качестве скринингового теста на выявление избыточной секреции кортизола, но для подтверждения требуется по крайней мере один дополнительный тест (определение свободного кортизола в суточной моче или ночной тест подавления с дексаметазоном). Расположение микроаденомы обычно определяется с помощью МРТ, хотя в сложных случаях может потребоваться селективный забор крови из нижних каменистых синусов. Транссфеноидальная хирургия является методом выбора при болезни Кушинга у детей. Сообщается о первичной ремиссии у 70-98% пациентов, а в долгосрочной перспективе у 50-98%. После операции часто наблюдается резидуальный транзиторный гипокортицизм, сохраняющийся в течение 30 мес. Если уровень кортизола остается повышенным и/или уровень АКТГ остается определяемым, используется лучевая терапия аденомы гипофиза. Успешное лечение может не привести к восстановлению роста, а впоследствии может возникнуть дефицит гормона роста, который следует лечить по мере необходимости.