а) Фолиевая кислота. Педиатров просят принять во внимание, что в развитии некоторых заболеваний задействованы эпигенетические механизмы. Предполагается, что эпигенетические процессы, обычно определяемые как регуляторный контроль экспрессии генов, способны перекрывать информацию, закодированную в последовательности ДНК, что выражается в увеличении или уменьшении риска развития заболевания. Несмотря на существование мощных геномных тестов для исследования этих регуляторов экспрессии генов, трудно четко ответить на вопрос, как эпигенетические механистические теории могут улучшить качество МП, оказываемой пациентам.
Уточнение основных понятий и определений, лежащих в основе предлагаемого эпигенетического вклада в фенотипы, должно привести к пониманию важности их роли в здоровье человека.
б) Эпигенетические механизмы заболеваний - моделирование на золотистых мышах из линии Viable Yellow. Эпигенетика (в буквальном переводе означает «над последовательностью ДНК»; «эпи» — над; «генетика» — последовательность ДНК) означает, что информация, закодированная в последовательности ДНК, м.б. изменена каким-либо образом с помощью информации более высокого порядка, которая регулирует уровни активности определенных генов.
Такая концепция заманчива, когда мы пытаемся понять, почему монозиготные близнецы, имеющие идентичные последовательности ДНК, иногда подвержены разл. риску возникновения определенных наследственных заболеваний, напр. болезни Альцгеймера или СД-1. Генетическая предрасположенность, которая не может полностью объяснить развитие заболевания или др. фенотипа, была названа «отсутствующей наследуемостью» — пробел, роль в восполнении которого было предложено возложить на эпигенетические регуляторные процессы.
Более того, поскольку окружающая среда влияет на риск возникновения определенных заболеваний, изменяя лежащую в их основе генетическую предрасположенность, факторы окружающей среды могут действовать через эпигенетические регуляторные процессы экспрессии генов.
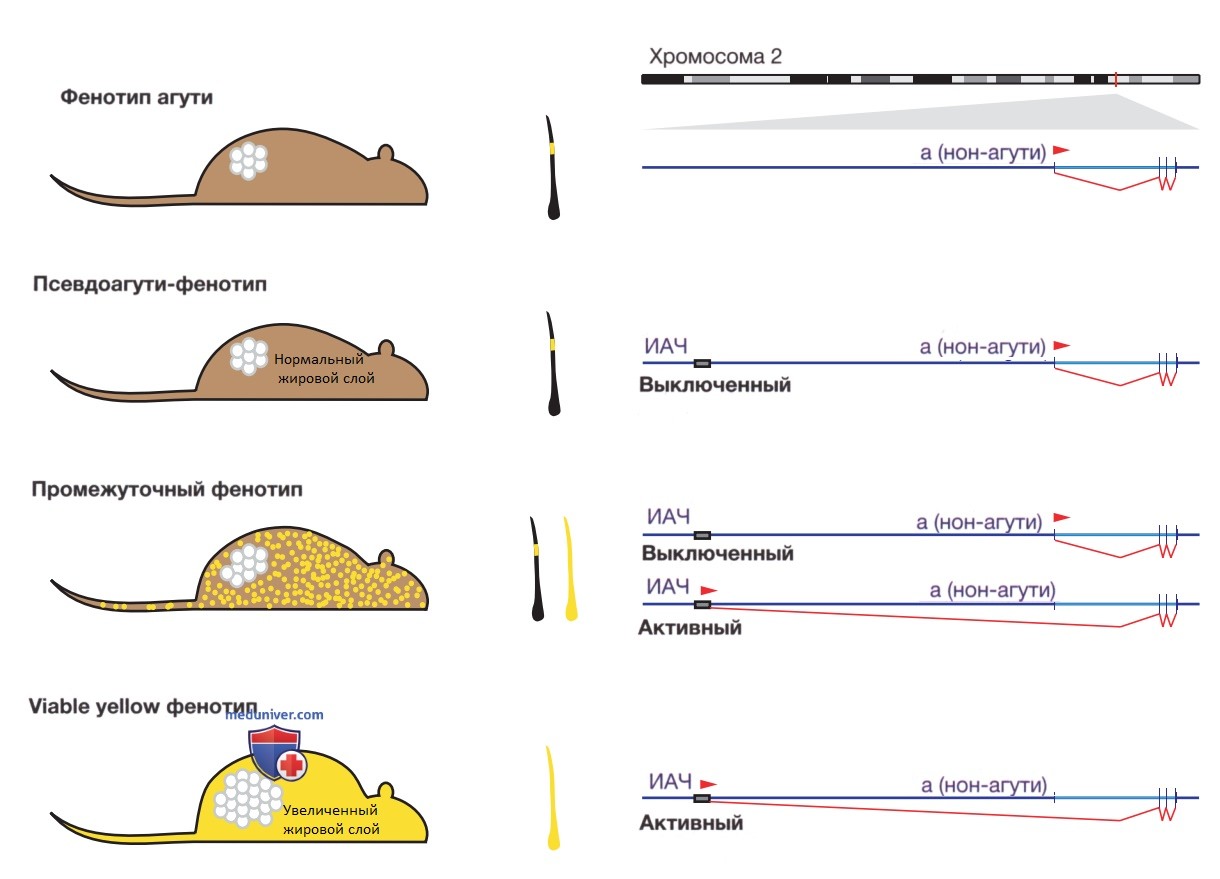
Наиболее убедительным доказательством наличия более высокого уровня эпигенетической регуляции генов и предрасположенности к заболеваниям стало моделирование на золотистых мышах из линии viable yellow (рис. 1). У этих мышей была обнаружена мутация, обусловленная внедрением эндогенного ретровируса — компонента генома, который мог реплицироваться и перемещаться на новую локацию. В случае с рыжими мышами viable yellow эндогенный ретровирус являлся так называемой интрацистернальной А-частицей (ИАЧ), инсерцированной перед геном а (нон-агути).
Рисунок 1. Модель эпигенетического изменения риска заболевания на модели золотистых мышей viable yellow. Штамм дикого типа изображен вверху; коричневый цвет шерсти вызван полоской желтого феомеланина внутри черного волосяного стержня, что является результатом всплеска активности экспрессии гена а (нон-агути) на хромосоме 2 во время роста шерсти. Примеры ниже иллюстрируют процесс, при котором интрацистернальный мобильный элемент А-частицы (ИАЧ) вставляется перед геном нон-агути. Такие мыши м.б. не отличимы от мышей дикого типа, если ИАЧ полностью выключен (псевдоагути-фенотип) или в случае активности ИАЧ в каждой кл. (внизу) он, управляя непрерывной транскрипцией гена нон-агути, стимулирует экспрессию феомеланина на всем протяжении роста волоса, чем обусловливает золотистый цвет шерсти (фенотип viable yellow). Эти мыши также страдают ожирением вследствие воздействия сигнального белка агути на адипоциты. Когда одни кл. экспрессируют, а др. выключают ИАЧ, образуется промежуточный фенотип шерсти, часто описываемый как «пятнистый», сопровождающийся менее выраженным ожирением. Это показывает, как одна и та же генетическая мутация (инсерция ИАЧ) изменчива в своей ассоциации с фенотипом в зависимости от различий в регуляции транскрипции в определенном локусе генома
Ген нон-агути кодирует предшественник сигнального белка агути, который связывается с рецепторами меланокортина и оказывает на них «-» эффект. Когда он стимулирует меланоциты в волосяных фолликулах, он вызывает выработку желтого пигмента феомеланина, а не черного эумеланина. Без ИАЧ, инсерцированной перед геном, нон-агути обычно включается на очень короткий момент и стимулирует выработку ограниченного количества желтого пигмента.
Было обнаружено, что присутствие активной ИАЧ перед геном создает новый, конститутивно активный стартовый кодон для гена нон-агути, что приводит к образованию шерстинок с феомеланином по всей их длине и, соответственно, характерной рыжей окраске фенотипа. Поскольку предшественник сигнального белка агути также экспрессируется и в др. типах кл., дополнительная активность гена нон-агути, управляемая ИАЧ, вызвала ожирение у золотистых мышей (вследствие воздействия на адипоциты), что послужило причиной развития у этих животных синдрома, сопоставимого с СД-2.
Мыши данного вида стали любопытной моделью потенциальной эпигенетической роли в риске возникновения заболевания вследствие неожиданного наблюдения, что детеныши из одного помета, все носители одной и той же инсерционной мутации ИАЧ, разительно различались по количеству золотистой шерсти и сопутствующему ожирению взрослой особи. У некоторых мышей было настолько мало золотистых шерстинок, что они практически не проявляли видимых признаков мутации.
ИАЧ у этих однопометников была активна в кл. золотистых мышей, как и ожидалось, но была выключена у особей с коричневой шерстью. Неактивная ИАЧ отличалась приобретенным ДНК-метилированием — модификацией цитозинов, расположенных непосредственно перед гуанинами (динуклеотиды CG или CpG), в 5-метилцитозин. Метилирование цитозинов в CG-динуклеотидах является пассивным состоянием генов во всем геноме, но обычно отсутствует в областях, регулирующих экспрессию близлежащих генов, поэтому его приобретение на этих участках указывает на то, что ген подвергся выключению.
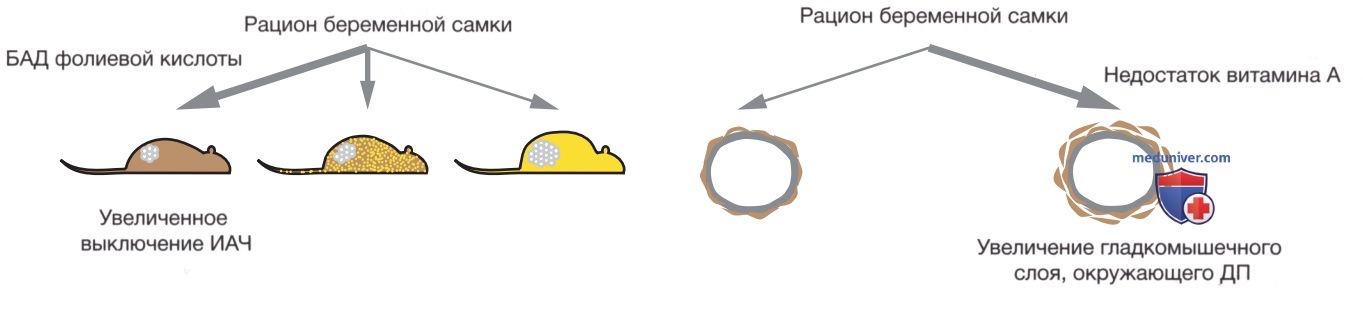
Этот факт позволяет предположить, что влияние на характер экспрессии генов притупляет врожденную генетическую предрасположенность, изменяя риск развития болезни. Кроме того, исследователи изменили диету беременных особей (детеныши в помете были носителями инсерции ИАЧ), добавив фолиевую кислоту — одноуглеродный донор, повысивший доступность кофактора, необходимого для метилирования ДНК. В результате у более высокого процента родившихся мышей наблюдалось ДНК-метилирование и инактивация мутации ИАЧ (рис. 2).
Рисунок 2. Модификация риска возникновения заболевания у взрослых особей вследствие изменения диеты беременной самки. Слева приведены псевдоагути-, промежуточные и viable yellow фенотипы, обусловленные различиями в активности интрацистернальной А-частицы (ИАЧ), инсерцированной перед геном нон-агути. Увеличение дозы фолиевой кислоты в рационе самки во время беременности приводит к увеличению количества детенышей, рожденных с псевдоагути- и промежуточным фенотипами, а также к меньшей степени выраженности ожирения во взрослом возрасте. Справа показан эффект нехватки витамина А в рационе беременной самки во время формирования легких у потомства. Недостаток витамина А во время развития приводит к увеличению объема гладких мышц (коричневый) вокруг дыхательных путей (ДП) и повышенному сопротивлению ДП в зрелом возрасте. Дефицит витамина А вызывает нехватку ретиноевой кислоты в развивающихся эмбрионах и, как следствие, нарушения регуляции экспрессии генов, которые изменяют метаболический путь кл., стимулируя образование большего количества гладкомышечных кл. Оба примера иллюстрируют, как влияние диеты во время беременности изменяет направление развития кл. путем регуляции экспрессии генов и предрасположенности к заболеваниям, проявляющимся во взрослом возрасте
Следовательно, причиной изменчивости развития у мышей золотистой окраски шерсти и ожирения м.б. влияние, которому подверглась самка во время беременности, напр. со стороны рациона. Этот факт подтвердил предположение, что в/утробный стресс приводит к повышению риска возникновения определенных заболеваний у взрослых, таких как ССЗ, нефрологические и метаболические заболевания. Эта область исследований, часто именуемая концепцией первопричин здоровья и болезней на ранних периодах развития, задается вопросом, как кл. человека помнят в/утробный стресс спустя годы или десятилетия.
Модель мышей с мутацией viable yellow позволяет предположить, что такая память м.б. опосредована регуляторами экспрессии генов и находиться под влиянием факторов окружающей среды, таких как питание матери во время беременности.
в) Эпигенетика и регулирование экспрессии генов. Два примера регуляции генов иллюстрируют модель блокирования системы регуляции на раннем этапе развития, а также ее дальнейшее сохранение на неопределенный срок. Первый — это инактивация Х-хромосомы. Поскольку мужчины имеют только одну Х-хромосому, она не подвергается инактивации. Однако индивид с двумя Х-хромосомами инактивирует одну, индивид с трисомией по Х-хромосоме инактивирует две и т.д.
В результате у мужчин и женщин имеется по одной активной Х-хромосоме на кл., несмотря на то что их развитие начинается с разного количества Х-хромосом. Этот процесс называется дозовой компенсацией. Инактивация Х-хромосомы, как правило, является случайным событием, при котором материнская Х-хромосома инактивируется в 1/2 кл. тела, а отцовская Х-хромосома — в др. 1/2. Инактивация происходит на ранней стадии развития организма при имплантации бластоцисты в стенку матки. Однако, единожды установившись в этом небольшом количестве плюрипотентных кл., характер инактивации сохраняется во всех кл. индивидуума на протяжении всей жизни.
Др. релевантной моделью регуляции генов является геномный импринтинг. Активация генов в кл. определенного типа обычно включается на копиях, присутствующих как на отцовской, так и на материнской хромосомах. Однако импринтированный локус отличается от остальных, потому что для некоторых импринтированных генов включается только копия, находящаяся либо на материнской, либо на отцовской хромосоме. Эта инактивация случается даже раньше, чем инактивация Х-хромосомы, происходящая во время образования мужских или женских гамет. Повторим, что эти модели инактивации сохраняются на протяжении всей жизни до старости.
- Эволюция термина «эпигенетика». Поскольку оба предыдущих примера включали процессы регуляции генов (выключение), происходившие на ранней стадии развития и сохранявшиеся в зрелом возрасте, они были описаны как «эпигенетические» с акцентированием внимания на то, каким образом кл. сохраняет память о прошлых регуляторных процессах. Т.о. подчеркивалась важность давно существовавшего мнения, что эпигенетика обладает вторым св-вом — опосредование клеточной памяти.
В 1950-х гг. Nanney интерпретировал эпигенетический ландшафт, чтобы дать определение эпигенетике как св-ву кл. запоминать прошлые события. В 1970-х гг. Riggs и Holliday отметили, что паттерны метилирования ДНК могут передаваться от родительских к дочерним кл., потенциально обеспечивая молекулярный механизм клеточной памяти; они описали это как «эпигенетическое св-во». Обнаружение факта, что метилирование ДНК является особенностью аллелей, выключаемых во время инактивации Х-хромосомы и геномного импринтинга, по всей вероятности, подтверждает идею о «наследуемой молекулярной метке», обусловливающей запоминание прошлого эпизода выключения во время развития организма, что позволяет описать метилирование ДНК как «эпигенетический регулятор».
В процессе дополнительного изучения активных и выключенных аллелей в Х-инактивированных или импринтированных локусах были обнаружены различия в состояниях хроматина и длинные некодирующие РНК, позволяющие различать хромосомы, и предположили, что они помогли обеспечить долгосрочное выключение этих локусов.
Были предприняты попытки проверить, наследуются ли состояния хроматина через деление кл. так же, как метилирование ДНК. Несмотря на то что доказательства их наследуемости представлялись менее убедительными, в отношении обозначения регуляторов транскрипции как эпигенетических — эта область имела тенденцию быть инклюзивной, а не эксклюзивной — потребовалось переопределить понятие эпигенетики как «эпи» (выше, над) и «генетика» (последовательность ДНК) в буквальном переводе. Это определение не только расходится с первоначальными представлениями о клеточном программировании и клеточной памяти, но и в настоящее время также включает в себя все процессы регуляции транскрипции.
Из-за расширенного определения эпигенетики эксперимент по проверке различий в клеточной памяти по своей методологии не отличается от эксперимента по проверке различий в регуляции транскрипции, которая может опосредовать или не опосредовать клеточную память.
г) Детские болезни с вовлечением эпигенетических процессов. Основными примерами эпигенетических изменений являются те, которые вызваны импринтированны-ми локусами, в частности синдромы Прадера-Вилли и Ангельмана. Каждый из этих синдромов м.б. обусловлен одной и той же делецией на хромосоме 15, отличаясь лишь тем, что делеция, произошедшая на отцовской хромосоме 15, приводит к развитию синдрома Прадера-Вилли и на материнской хромосоме 15 — к синдрому Ангельмана. Существуют импринтированные гены, расположенные в области 15q11-q13, некоторые из них экспрессируются только на отцовской хромосоме, в то время как другие — только на материнской.
При отсутствии участка на отцовской унаследованной хромосоме у человека все еще есть копия гена на оставшейся материнской хромосоме, но, если она выключается импринтингом, у него фактически нет функциональной копии гена, что приводит к формированию фенотипа Прадера-Вилли. Обратное происходит при синдроме Ангельмана; делеция материнской хромосомы заставляет «замолчать» копию гена на отцовской хромосоме.
Хотя причина развития этих синдромов у большинства носителей — делеции, у некоторой части пациентов они являются следствием однородительской дисомии (ОРД), при которой обе хромосомы 15 интактны, но обе наследуются от одного родителя. Материнская ОРД имеет тот же эффект, что и отцовская делеция, поскольку в ней отсутствует отцовская наследуемая хромосома, обусловливающая развитие синдрома Прадера-Вилли, а отцовская ОРД приводит к синдрому Ангельмана. Считается, что ОРД начинается с трисомии по этой хромосоме, за которой на ранней стадии развития следует потеря одной из трех хромосом, при этом иногда остаются две хромосомы, происходящие от одного и того же родителя. В дальнейшем у очень небольшой части лиц мутации в области 15q11—q13, по всей вероятности, влияют на импринтируемый домен в целом.
Синдромы Прадера-Вилли и Ангельмана возникают из-за генетических мутаций: больших делеций, случаев нерасхождения, ведущих к приобретению или потере целых хромосом, или меньших мутаций ДНК. При данных мутациях обнаруживается обусловливающая их форма геномного импринтинга — характерная организация генной регуляции на хромосоме 15q11-q13, которая отражает память о происхождении гаметы каждой хромосомы, описываемую как эпигенетическую. Чего не происходит у этих индивидуумов, так это изменения нормальной эпигенетической регуляции локуса, как на модели золотистых мышей агути. Чтобы выявить случаи нарушенной эпигенетической регуляции, приводящей к развитию заболевания, исследователи прибегают к тестам, изучающим формы метилирования ДНК по всему геному.
Если бы мы, напр., никогда не знали об инсерции ИАЧ у золотистых мышей агути, локус обнаружил бы себя по характерному метилированию ДНК у золотистых животных с ожирением при их сравнении с темношерстными генетически идентичными однопометниками с нормальной МТ. Этот метод, называемый полноэпигеномным исследованием ассоциаций (EWAS; англ. epigenome-wide association study), первоначально применялся для пациентов с в/утробными нарушениями, подвергшихся вредному воздействию окружающей среды или страдающих от разл. форм ЗНО, для поиска процессов клеточного репрограммирования.
д) Исследования эпигеномных ассоциаций - метилирование дезоксирибонуклеиновой кислоты. Благодаря доступности полногеномных анализов и продемонстрированной наследуемости через митоз, метилирование ДНК было основным направлением исследований, пытающихся установить связь между нарушениями эпигенетической регуляции и фенотипами заболевания. Схема полногеномных исследований ассоциаций (GWAS; англ. genome-wide association study) была использована для исследований EWAS, но вместо изменчивости последовательностей ДНК метод EWAS устанавливает связь между изменчивостью метилирования ДНК и фенотипом.
Интерпретация исследований EWAS является более сложной, чем предполагалось, отчасти потому, что метилирование ДНК в образце кл. отражает не только перепрограммирование, происходящее в этих кл., но и др. влияния. Напр., если образец кл. содержит >2 подтипов (каждый подтип кл. в теле человека имеет характерные формы метилирования ДНК), изменение пропорций подтипов кл. у людей вызовет изменение форм метилирования ДНК в локусах, где различия в метилировании ДНК характеризуют подтипы кл. Т.о., изменения метилирования ДНК м.б. обнаружены методом EWAS без изменения отдельными кл. их ДНК-метилирования.
Др. важным влиянием, на которое приходится 22-80% вариаций метилирования ДНК у человека, является вариабельность последовательности ДНК. Под эпигенетической регуляцией понимают уровень информации, превосходящий таковой у последовательности ДНК, но важность взаимного влияния вариабельности последовательности ДНК на метилирование ДНК весьма существенна. При интерпретации результатов стандартных исследований EWAS генетические вариации до настоящего времени во внимание не принимались, что в очередной раз предполагало неправильную интерпретацию изменений метилирования ДНК как отражающих репрограммирование кл., хотя на самом деле различия в метилировании ДНК могут отражать различия в последовательностях ДНК у исследуемых лиц.
Типичный алгоритм стандартного метода EWAS — одномоментное поперечное исследование: сравнение группы лиц с заболеванием и группы лиц, не затронутых этим заболеванием; к моменту начала исследования у данных лиц уже развилось интересующее исследователей заболевание. Это делает исследование уязвимым для эффектов обратной причинно-следственной связи, когда изучаемое заболевание изменяет метилирование ДНК, а не перепрограммирование метилирования ДНК вызывает заболевание. Было показано, что это происходит в лейкоцитах периферической крови у лиц с высоким ИМТ. Поэтому результаты всех исследований EWAS на сегодняшний день следует интерпретировать с осторожностью.
е) Вариабельность клеточного программирования как модель для эпигенетики и заболевания. Существует общепринятое мнение, что один или несколько типов кл. в организме претерпевают изменение в регуляции экспрессии генов — тип клеточного перепрограммирования, изменяющий св-ва кл. и способствующий развитию болезни. Однако др. модель, которую следует принять во внимание, — это перепрограммирование, происходящее раньше, в процессе программирования и изменения программы кл. в органе т.о., что это предрасполагает к заболеванию.
Исследования проводятся на мышиной модели, характеризующейся дефицитом витамина А у беременной самки. Витамин А является диетическим предшественником ретиноевой кислоты, который связывается с рецептором ретиноевой кислоты, впоследствии попадая в определенные точки генома для регуляции экспрессии групп генов. Когда рацион беременных самок мышей был ограничен по содержанию витамина А с 9,5-14,5 дня эмбрионального развития — период интенсивного формирования легких, — мыши рождались с увеличенным количеством гладких мышц, окружающих ДП, а позже, в процессе их жизни, было доказано, что это приводит к повышению сопротивления ДП.
Т.о., мыши демонстрировали компонент фенотипа реактивного заболевания ДП (БА), вызванный исключительно дефицитом питательного микроэлемента во время в/утробной жизни (см. рис. 2). Изменение клеточным программированием пропорций >1 типа кл. в зрелом органе является удивительно заманчивым механизмом для концепции первопричин здоровья и болезней на ранних периодах развития; это хорошо согласовывается с исходным определением эпигенетических событий, основанных на клеточном программировании, но не рассматривается как представляющий интерес результат в текущих исследованиях EWAS.
Та же самая модель м.б. рассмотрена для эпигенетического ответа на токсины, в частности на эндокринные разрушители, определяемые по их взаимодействию с эндокринной системой, для которых часто пытались обнаружить связь с эпигенетическими регуляторными процессами. Один интересный класс хим. соединений, разрушающих эндокринную систему, — это органотины — оловоорганические соединения, биоциды, используемые на разл. производствах. Было обнаружено, что органотин трибутилолово является причиной ожирения и направляет дифференцировку тканевых стволовых кл. преимущественно в сторону адипоцитов, передавая сигналы через транскрипционный путь с участием PPAR-γ.
Повторим: это не результат, к которому в настоящее время стремятся в обычных исследованиях EWAS, но он отражает нарушения, обусловливающие изменение клеточного программирования через механизмы регуляции транскрипции и приводящие к изменению клеточного состава у экспонированного индивидуума.
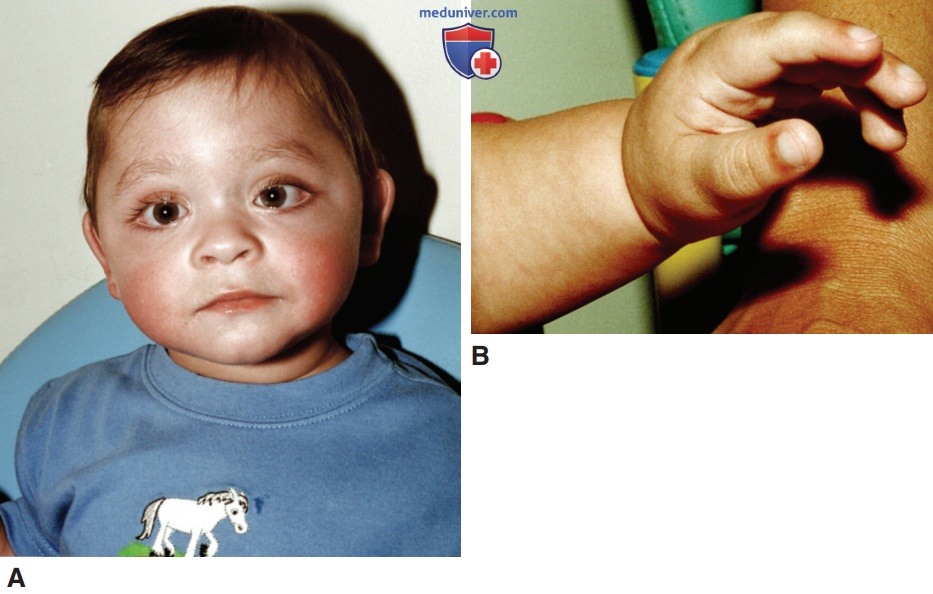
ж) Эпигенетические заболевания и терапевтические вмешательства. Возникает вопрос, могут ли терапевтические вмешательства улучшить или обратить вспять фенотип заболевания, если он возник вследствие эпигенетических процессов. При ЗНО, обусловленных соматическими мутациями, способными таргетировать разл. медиаторы регуляции транскрипции, появились многочисленные перспективные терапевтические возможности. Интересным примером доброкачественной патологии является генетическое заболевание, называемое синдромом Кабуки, которое вызвано мутациями гена гистоновой метилтрансферазы (KMT2D) или гистоновой деметилазы (KDM6A), каждый из которых играет роль в создании доступного хроматина, обусловливающего надлежащую экспрессию соответствующих генов (рис. 3, табл. 1).
Рисунок 3. Синдром Кабуки у мальчика 18 мес.: А — длинные глазные щели и эктропион (выворот) нижних век; В — выступающие подушечки пальцев
С целью подтверждения гипотезы о том, что увеличение количества ацетилирования гистонов может помочь компенсировать несоответствующее метилирование гистонов, мышей с синдромом Кабуки подвергли приему кетогенной диеты, которая стимулирует увеличение образования β-гидроксибутирата — эндогенного ингибитора деацетилаз гистонов. У мышей, находящихся на этой диете, улучшился нейрогенез и память; это позволяет предположить, что такое вмешательство у детей с синдромом Кабуки также может иметь «+» эффекты.