а) Однородительская дисомия. Однородительская дисомия возникает, когда обе хромосомы из пары / участки одной хромосомы унаследованы от одного родителя. Однородительская дисомия м.б. двух типов: монородительская изодисомия/монородительская гетеродисомия. Однородительская изодисомия означает, что обе хромосомы/хромосомные области идентичны (обычно в результате процесса коррекции моносомии путем дупликации). Однородительская гетеродисомия означает, что пара неидентичных хромосом унаследована от одного родителя.
Это происходит в результате трисомии, которая позже превращается в дисомию, в результате чего от одного родителя остаются две копии. Фенотипический результат однородительской дисомии варьирует в зависимости от вовлеченной хромосомы родителя и от того, является ли это изодисомией/гетеродисомией. При однородительской дисомии наблюдаются три типа фенотипических эффектов: связанные с импринтированными генами (т.е. отсутствие гена, который обычно экспрессируется только при наследовании от родителя определенного пола), связанные с выявлением АуР-расстройств, а также связанные с рудиментарной анеуплоидией, обусловливающей возникновение мозаицизма.
При однородительской изодисомии обе хромосомы/области (следовательно, и гены) в паре идентичны. Это особенно важно, когда родитель является носителем АуР-заболевания. Если у потомка родителя-носителя имеется однородительская дисомия с изодисомией для хромосомы, несущей аномальный ген, аномальный ген будет присутствовать в двух копиях, и фенотип будет фенотипом АуР-расстройства; у ребенка разовьется АуР-заболевание, хотя только один родитель является носителем этого рецессивного заболевания.
По предварительным подсчетам, все люди являются носителями примерно 20 аномальных АуР-генов. В случаях однородительской дисомии сообщалось о некоторых АуР-расстройствах, таких как СМА, кистозный фиброз, гипоплазия волос и хрящей, α-/β-талассемии и синдром Блума. Возможность однородительской изодисомии также следует учитывать, когда человек страдает >2 рецессивными заболеваниями, поскольку аномальные гены обоих заболеваний могут находиться на одной и той же изодисомной хромосоме. Однородительская изодисомия редко является причиной рецессивно наследуемых заболеваний. Однородительские изодисомы также можно обнаружить с помощью микрочипов SNP.
Материнские однородительские дисомии по хромосомам 2, 7, 14 и 15 и отцовские однородительские дисомии по хромосомам 6, 11, 15 и 20 связаны с фенотипическими нарушениями роста и поведения. Однородительская дисомия материнской хромосомы 7 обусловливает фенотип, аналогичный синдрому Рассела-Сильвера с задержкой в/ утробного развития. Эти фенотипические эффекты могут быть обусловлены импринтингом (см. ниже) (рис. 1).
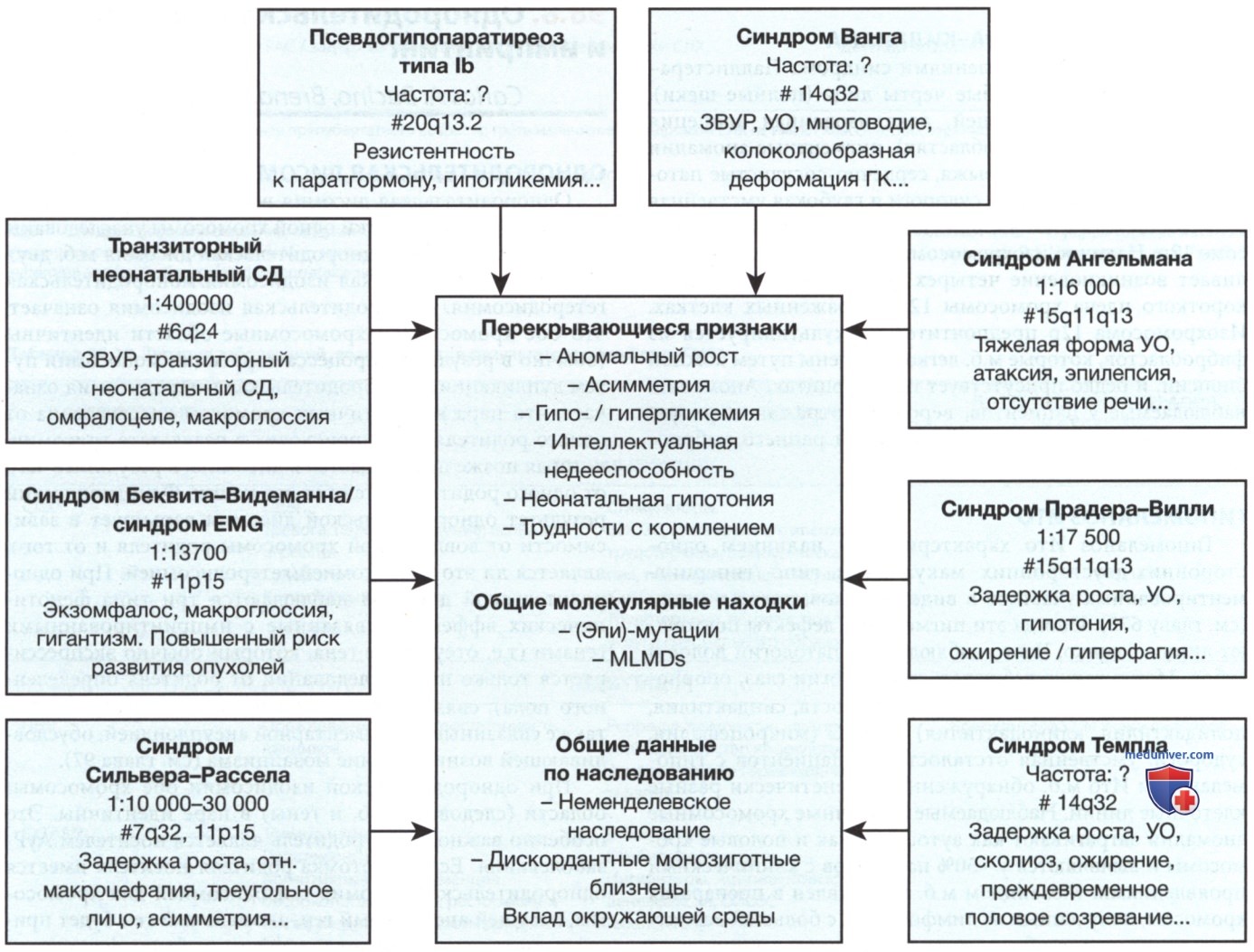
Рисунок 1. Обзор общих результатов клинических и молекулярных анализов при восьми наиболее часто встречающихся импринтинговых заболеваниях, их специфические особенности, частота возникновения и основная хромосомная локализация. #6q24 — хромосома 6q24; EMG — эк-зомфалоз (Е), макроглоссия (М) и гигантизм (G), ЗВУР — задержка в/утробного развития; УО — умственная отсталость; MLMDs — мультилокусные дефекты метилирования
Однородительская дисомия по хромосоме 15 наблюдается у некоторых пациентов с синдромом Прадера-Вилли и синдромом Ангельмана. У 25-29% пациентов с синдромом Прадера-Вилли обнаруживается материнская однородительская дисомия (отцовская хромосома 15 отсутствует) (рис. 2). При синдроме Ангельмана отцовская однородительская дисомия по хромосоме 15 встречается реже и наблюдается в 5% случаев (отсутствует материнская хромосома 15). Считается, что фенотип синдрома Прадера-Вилли и синдрома Ангельмана в случаях однородительской дисомии возникает в результате отсутствия функционального вклада со стороны конкретного родителя (хромосомы 15). При синдроме Прадера-Вилли отсутствует отцовский вклад, а при синдроме Ангельмана — материнский вклад.
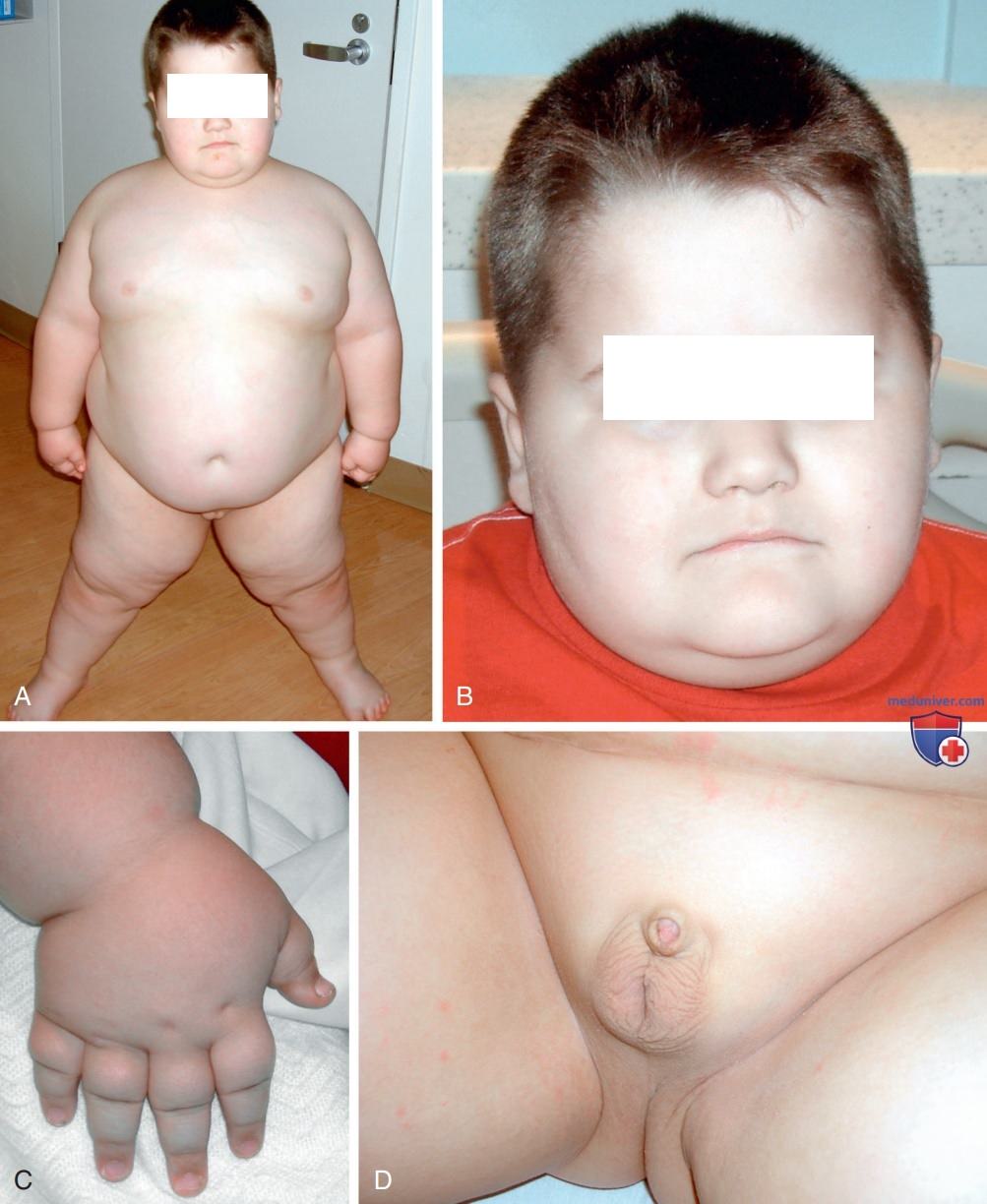
Рисунок 2. Фенотип Прадера-Вилли: А, В — ребенок с патологическим ожирением и характерными чертами лица; C — верхние конечности отличаются маленьким размером кистей по сравнению с размером тела; D — наружные гениталии после лапароскопической орхиопексии в 13 мес. На публикацию фотографий было получено информированное согласие родителей, одобренное наблюдательным советом медколледжа Бейлора
Синдром Прадера-Вилли м.б. вызван отцовской недостаточностью НВ11-85 — малых ядрышковых РНК (snoRNAs). Эти данные позволяют предположить существующие различия в функциях определенных участков хромосомы 15 в зависимости от того, унаследована ли она от матери/отца. Синдром Ангельмана вызван отсутствием материнского гена, известного как UBE3A, и м.б. результатом материнской делеции, материнской мутации UBE3A, отцовской однородительской дисомии и аномалий в материнском центре импринтинга в области хромосомы 15q11-13.
Однородительская дисомия чаще всего возникает, когда беременность начинается с трисомического зачатия, за которым следует процесс «коррекции трисомии». Поскольку большинство трисомий приводит к летальному исходу, плод может выжить только в том случае, если линия клеток потеряет лишнюю хромосому и вернется в дисомное состояние. В 1/3 случаев линия дисомных клеток является однородительской. Это классический механизм синдрома Прадера-Вилли, который возникает чаще с увеличением возраста матери. Эмбрион начинает развиваться с трисомией 15, вторичной по отношению к не-расхождению в материнском мейозе I, за которым следует случайная потеря отцовской хромосомы. В этом случае линия дисомных клеток приобретает большую жизнеспособность и перерастает линию трисомных клеток.
Когда мозаичная трисомия обнаруживается во время пренатальной диагностики, следует позаботиться о выявлении однородительской дисомии, а также определить, является ли вовлеченная хромосома одной из дисомий, связанных с фенотипическими аномалиями. Всегда существует опасение, что некоторые остаточные клетки, являющиеся трисомными, присутствуют в некоторых тканях, что приводит к порокам/дисфункции. Наличие агрегатов трисомных клеток может объяснить спектр патологий, наблюдаемых у людей с однородительской дисомией.
б) Импритинг. Традиционная генетика на протяжении многих лет предполагала, что большинство генов одинаково экспрессируются при наследовании по материнской и отцовской линии. Единственным исключением из этого правила являлись гены на Х-хромосоме, подлежащие инактивации, и гены Ig, подверженные аллельному исключению, — феномен, который приводит к моноаллельной экспрессии конкретной цепи Ig путем включения и выключения экспрессии родительских аллелей.
Геномный импринтинг имеет место, когда фенотипическая экспрессия гена зависит от родительского происхождения определенных генов или, в некоторых случаях, целых участков хромосомы. Экспрессируется ли генетический материал или нет, зависит от пола родителя, от которого он был получен. В некоторых случаях геномный импринтинг можно заподозрить на основании родословной. В подобных родословных наследственная болезнь всегда проявляется только у представителей одного пола, являясь клинически «молчащей» у противоположного пола на протяжении нескольких поколений (рис. 3, 4).
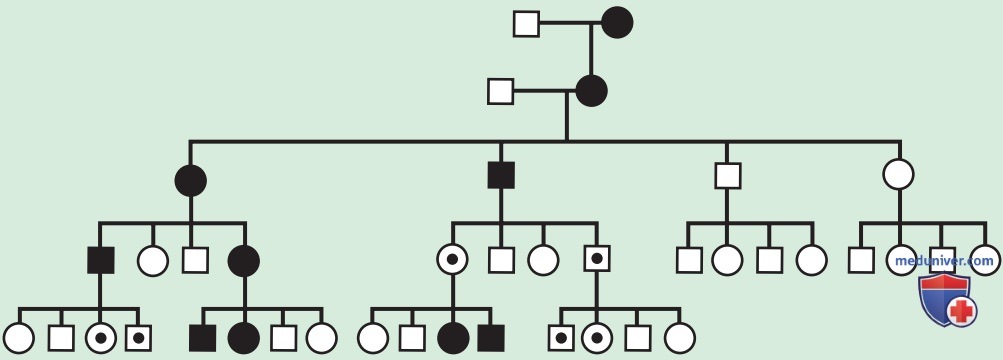
Рисунок 3. В этой гипотетической родословной, предполагающей импринтинг, фенотипические эффекты проявляются только тогда, когда мутировавший ген передается от матери, а не от отца, т.е. имеет место материнская недостаточность. Равное количество мужчин и женщин могут иметь / не иметь фенотипические проявления в каждом поколении. Бессимптомный трансмиттер дает ключ к определению пола родителя, который передает экспрессированную генетическую информацию; т.е. при болезнях, вызванных материнской недостаточностью (также называемых отцовским импринтингом), существуют «пропущенные» бессимптомные женщины. Это теоретическое рассуждение, потому что в большинстве клинических сценариев материнской недостаточности, таких как синдром Ангельмана, люди с данной патологией не имеют потомства
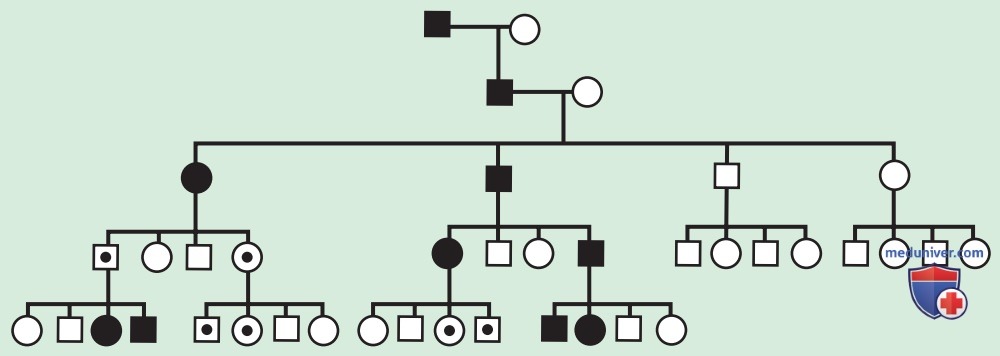
Рисунок 4. В теоретических родословных, предполагающих отцовскую недостаточность (материнский импринтинг), фенотипические эффекты возникают только тогда, когда мутировавший ген передается от отца, а не от матери. Равное количество мужчин и женщин может иметь / не иметь фенотипические проявления в каждом поколении. В теоретической ситуации бессимптомный трансмиттер дает ключ к пониманию пола родителя, который передает экспрессированную генетическую информацию; т.е. при отцовской недостаточности (также известной как материнский импринтинг) существуют «пропущенные» бессимптомные мужчины
По всей видимости, импринтинг происходит во многих различных частях генома человека и считается особенно важным для экспрессии генов, связанных с развитием, ростом, возникновением онкологических заболеваний и даже поведением; >60 генов классифицируются как импринтируемые. Болезни импринтинга могут возникать в результате однородительской дисомии, делеций/дупликаций, эпигенетических паттернов аберрантного метилирования / точечных мутаций в конкретном гене.
Классическим примером болезней импринтинга являются синдром Прадера-Вилли и синдром Ангельмана — два очень разных клинических состояния. Эти синдромы обычно развиваются вследствие делеции одной и той же области в проксимальном длинном плече хромосомы 15. Делеция на отцовской хромосоме обусловливает возникновение синдрома Прадера-Вилли, при котором материнская копия не повреждена, но некоторые из импринтированных генов на этом участке обычно молчат. Синдром Прадера-Вилли можно диагностировать клинически (табл. 20) и подтвердить с помощью генетического тестирования. Дополнительные клинические особенности и проблемы увеличения МТ приведены в табл. 21.
Набор массы тела (МТ) трудно контролировать, но лечение гормоном роста привело к увеличению роста, мышечной массы, уменьшению объема жировой ткани и улучшению когнитивных функций.
Материнская делеция той же области, что и при синдроме Прадера-Вилли, обусловливает возникновение синдрома Ангельмана, оставляя нетронутой отцовскую копию, которая в этом случае содержит обычно молчащие гены. В др. ситуациях к тому же диагнозу может привести однородительская дисомия (табл. 22). Многие др. заболевания связаны с подобным эффектом родительского происхождения, напр. некоторые случаи синдрома Беквита-Видемана, синдрома Рассела-Сильвера и неонатальный СД.