а) Болезнь Пелицеуса-Мерцбахера. Болезнь Пелицеуса-Мерцбахера (Pelizaeus, Merzbacher) — это Х-сцепленное рецессивное расстройство, характеризующееся нистагмом и аномалиями миелина. Данная патология вызывается мутациями в гене протеолипидного белка (PLP1) на хромосоме Xq22, который необходим для образования миелина в ЦНС и дифференцировки олигодендроцитов. Мутации в одном и том же гене могут вызывать семейный спастический парапарез (прогрессирующий спастический парапарез типа 2).
Мутации PLP1, вызывающие заболевание, включают точечные мутации, делеции, дупликации и др. изменения генов.
Клинически классическая болезнь Пелицеуса-Мерцбахера диагностируется в младенчестве по нистагму и блуждающим движениям глаз, кивкам головы. Отмечается задержка развития; в последующем развиваются атаксия, хореоатетоз и спастичность. Атрофия зрительного нерва и дизартрия являются сопутствующими симптомами, смерть наступает на втором/третьем десятилетии жизни. Основа патогенеза — потеря миелина с интактными аксонами, что указывает на дефект функции олигодендроглии.
На МРТ выявляется картина симметричной замедленной миелинизации. В настоящее время признано, что широкий спектр фенотипов, включая прогрессирующий спастический парапарез и аномалии периферических нервов, м.б. результатом мутаций в гене PLP1.
Продолжают выявляться др. подобные данной патологии гипомиелинизирующие лейкодистрофии, что должно учитываться при ДД. К ним относятся синдром Аллана-Херндона-Дадли (Allan, Herndon, Dudley) и расстройства TUBB4A.
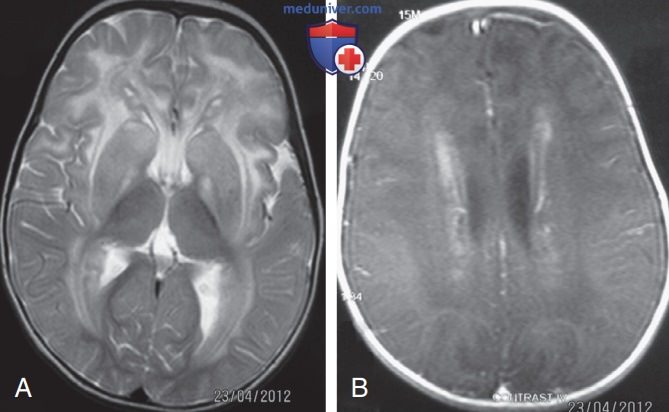
б) Болезнь Александера. Редкое заболевание, включающее прогрессирующую макроцефалию и лейкодистрофию. Болезнь Александера (Alexander) вызвана доминантными мутациями в гене глиального фибриллярного кислого белка (GFAP; англ. glial fibrillary acidic protein) на хромосоме 17q21, и случаи заболевания в семьях обычно спорадичны. Патологоанатомическое исследование ГМ выявляет отложение эозинофильных гиалиновых тел, называемых волокнами Розенталя (Rosenthal), в отростках астроцитов. Они накапливаются периваскулярно по всему ГМ.
При классической инфантильной форме болезни Александера дегенерация белого в-ва наиболее выражена фронтально. Диагноз м.б. поставлен по МРТ (рис. 1) и MP-спектроскопии, выявляющей патологические метаболические субстраты. У пострадавших детей развивается прогрессирующая потеря интеллекта, спастичность и резистентные приступы, приводящие к смерти к 5 годам. Однако есть более мягкие формы, которые появляются позже и могут не иметь характерной лобной локализации/мегалэнцефалии.
Рисунок 1. Болезнь Александера. Магнитно-резонансная томограмма пациента 15 мес: А — осевые Т2-взвешенные изображения (TR/TE: 4000/99) на уровне базальных ганглиев и таламуса, демонстрирующие диффузный двусторонний симметричный повышенный сигнал преимущественно лобного перивентрикулярного, но также подкоркового, белого в-ва и базальных ганглиев; В — значительный перивентрикулярный ободок после в/в инфузии гадолиния (Т1-взвешенные изображения; TR/TE: 400/88)
в) Губчатая дегенерация Канавана. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
г) Другие лейкодистрофии. Метаболические и дегенеративные нарушения могут сопровождаться значительными изменениями белого в-ва ГМ, такими как некоторые митохондриальные нарушения и глутаровая ацидурия типа 1. Кроме того, более широкое использование МРТ ГМ позволило выявить новые лейкодистрофии.
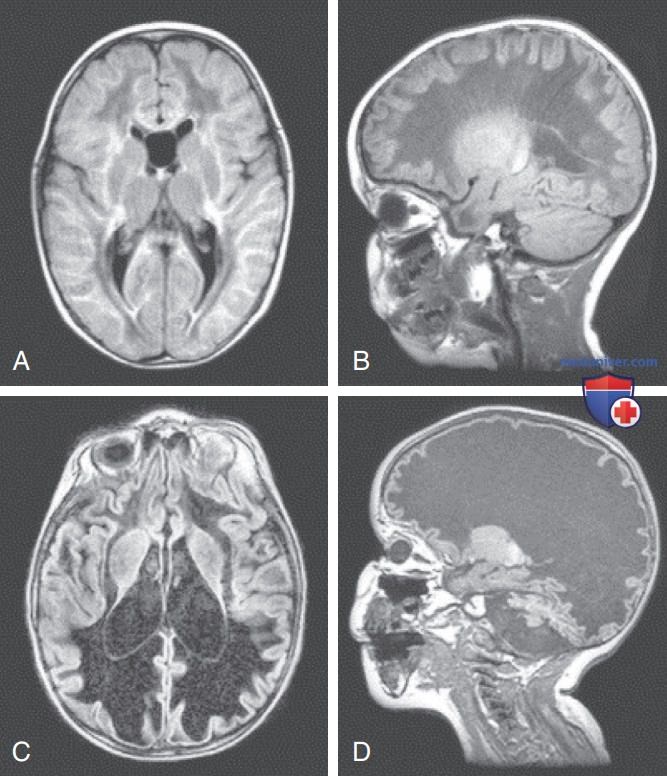
Одним из примеров является болезнь исчезающего белого в-ва/детская атаксия с гипомиелинизацией ЦНС, характеризующаяся атаксией и спастичностью (рис. 2). У некоторых пациентов наблюдаются атрофия зрительного нерва, судороги и снижение когнитивных функций. Возраст дебюта и скорость формирования лейкодистрофии м.б. весьма вариабельными. При ранней форме развитие обычно стремительно, что быстро приводит к смерти; при поздней форме снижение умственных способностей, как правило, происходит медленнее и мягче. Интересно, что острая демиелинизация при этих расстройствах м.б. вызвана лихорадкой/стрессом.
Рисунок 2. Т1-взвешенные и изображения в режиме восстановления инверсии с ослаблением жидкости пациента с болезнью исчезающего белого в-ва. Осевое восстановление инверсии с ослаблением жидкости (А, С) и сагиттальное Т1-взвешенное (В, D) изображения пациента 1,5 и 2,5 лет. Первая магнитно-резонансная томография (A, B) проведена вскоре после появления симптомов заболевания. Начальное изображение в режиме восстановления инверсии с ослаблением жидкости (A) демонстрирует диффузную аномалию и частичную кистозную дегенерацию белого в-ва головного мозга, тогда как последующее изображение в режиме восстановления инверсии с ослаблением жидкости (B) показывает, что все белое в-во головного мозга было заменено жидкостью. Исходное Т1-взвешенное сагиттальное изображение (B) выявляет типичный полосообразный рисунок внутри аномального белого в-ва, тогда как последующее изображение (C) показывает, что белое в-во головного мозга отсутствует и сохранились только кора и эпендимальная выстилка. Удивительно, но отсутствующее белое в-во выглядит отечным вследствие растяжения вышележащей коры в области широких извилин. Мозжечок значительно атрофирован.
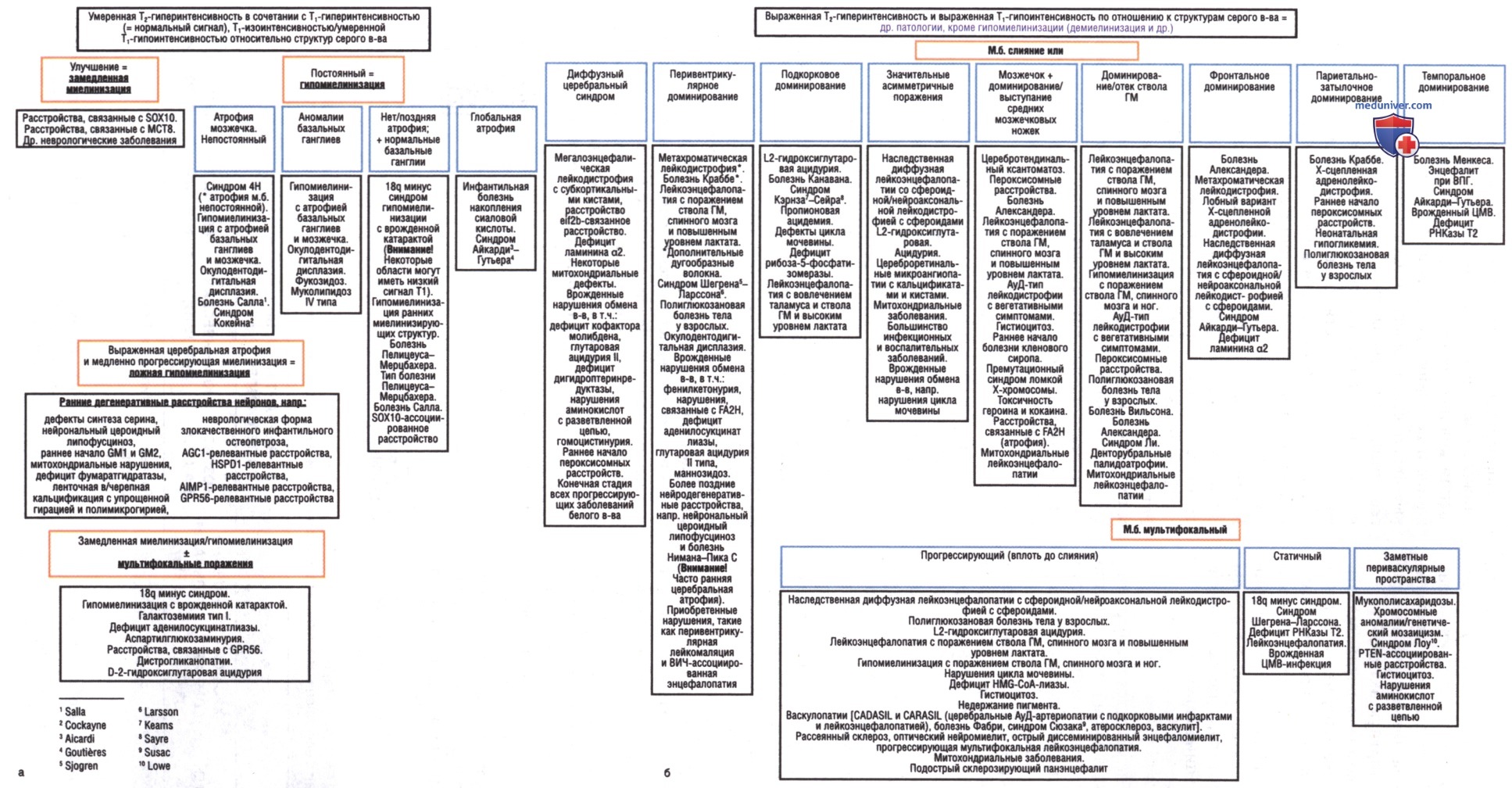
Диагноз болезни исчезающего белого в-ва/детской атаксии с гипомиелинизацией ЦНС основывается на клинических данных, характерных аномалиях на МРТ ГМ и АуР-мутациях в одном из пяти причинных генов (EIF2B1, EIF2B2, EIF2B3, EIF2B4 и EIF2B5), кодирующих пять субъединиц фактора инициации эукариотической трансляции (eIF2B). Диагностика лейкодистрофий, основанная на результатах МРТ, представлена на рис. 3, клиническая и лабораторная часть — в табл. 3.
Рисунок 3. Определение магнитно-резонансных паттернов при лейкодистрофиях и генетических лейкоэнцефалопатиях. Три основные характеристики магнитно-резонансной томографии помогают различать типы лейкодистрофии и лейкоэнцефалопатии. Первый дискриминатор — это наличие/отсутствие гипомиелинизации (а). В пределах этого подмножества наличие улучшения миелинизации/атрофии направляет клинициста к серии лейкоэнцефалопатий. При истинных гипомиелинизирующих лейкодистрофиях наличие базальных ганглиев и мозжечкового поражения дополнительно помогает уточнить диагноз. Если паттерн не связан с гипомиелинизацией, то второй дискриминатор заключается в том, являются ли аномалии белого в-ва слитными/изолированными и мультифокальными (б). Если аномалии белого в-ва сливаются, то третьим дискриминатором является преобладающая локализация аномалий (б). 4Н — гипомиелинизация, гиподонтия и гипогонадотропный гипогонадизм; НАСВ — гипомиелинизация с атрофией базальных ганглиев и мозжечка; HEMS — гипомиелинизация ранних миелинизирующих структур; ODDD — окулодентодигитальная дисплазия; НСС — гипомиелинизация с врожденной катарактой; PMD — болезнь Пелицеуса-Мерцбахера; PMLD — тип болезни Пелицеуса-Мерцбахера; NCL — нейрональный цероидный липофусциноз; APDB — полиглюкозановая болезнь тела у взрослых; ADLD — аутосомно-доминантный тип лейкодистрофии с вегетативными симптомами; CRMCC — цереброретинальные микроангиопатии с кальцификатами и кистами; СТХ — церебротендинальный ксантоматоз; DRPLA — денторубральные палидоатрофии; Е1F2В-связанные расстройства (исчезающего белого в-ва болезни/САСН); HDLS — наследственная диффузная лейкоэнцефалопатии со сфероидной/ нейроаксональной лейкодистрофией с сфероидами; HBSL — гипомиелинизация с поражением ствола головного мозга, спинного мозга и ног; LTBL — лейкоэнцефалопатия с вовлечением таламуса и ствола головного мозга и высоким уровнем лактата; LBSL — лейкоэнцефалопатия с поражением ствола головного мозга, спинного мозга и повышенным уровнем лактата; MLC — мегалоэнцефалическая лейкодистрофия с субкортикальными кистами; X-ALD — Х-сцепленная адренолейкодистрофия.
д) Болезнь Менкеса. Болезнь Менкеса (болезнь кудрявых волос) — прогрессирующее нейродегенеративное заболевание, наследуемое как Х-сцепленный рецессивный признак. Ген Менкеса — АТР7А — на Xq21.1, кодирует транспортирующую медь аденозинтрифосфатазу P-типа, и мутации в этом белке сопровождаются низким уровнем меди и церулоплазмина в сыворотке крови, дефектом всасывания и транспорта меди в кишечнике. Симптомы начинаются в первые месяцы жизни и включают гипотермию, гипотонию и генерализованные миоклонические приступы. Лицо с пухлыми, розовыми щеками и курчавыми, бесцветными, рыхлыми волосами.
Микроскопическое исследование волос показывает несколько аномалий, включая trichorrhexis nodosa (переломы вдоль стержня волоса) и pili torti (скрученные волосы). Трудности с кормлением приводят к задержке развития. Тяжелые когнитивные нарушения и атрофия зрительного нерва являются постоянными признаками заболевания. Морфологические изменения включают извилистую дегенерацию серого в-ва и выраженные изменения в мозжечке с потерей внутреннего гранулоцитарного слоя и некрозом клеток Пуркинье (Purkinje).
Смерть может наступить к 3 годам жизни при отсутствии лечения. Очень редко болезнь Менкеса поражает женщин, характерна более мягкая симптоматика.
Терапия гистидином меди м.б. эффективной в предотвращении неврологического ухудшения у некоторых пациентов с болезнью Менкеса, особенно когда лечение начинается в неонатальном периоде/предпочтительно, в период в/утробного развития. У детей в настоящее время возможна диагностика в пресимптомный период при наличии болеющего брата в семейном анамнезе. Медь необходима на ранних стадиях развития ЦНС, и ее отсутствие, вероятно, объясняет морфологические изменения.
Младенцам, с диагностированным пресимптомно заболеванием в первые 10 дней жизни, м.б. начато экспериментальное лечение по протоколу ежедневных п/к инъекций гистидина меди (по состоянию на 2017 г. доступны только в Национальном институте здоровья по программе, контролируемой доктором Стивеном Калером (Stephen Kaler)). Оптимальный ответ на лечение инъекциями гистидина меди, по-видимому, возникает только у пациентов с установленным в период новорожденности диагнозом и чьи мутации допускают остаточную медьтранспортную активность.
Синдром затылочного рога, скелетная дисплазия, вызванная разл. мутациями в том же гене, что и болезнь Менкеса, являются относительно легкими проявлениями. Эти два заболевания часто путают, т.к. биохимические аномалии идентичны. Принятие решения в отношении лечения пациентов с болезнью Менкеса требует тщательной корреляции генотипа и фенотипа, а также дальнейших клинических испытаний терапии медью.
е) Синдром Ретта. Этот синдром, строго говоря, не является дегенеративным заболеванием, а представляет собой расстройство раннего развития ГМ, отмеченное периодом регрессии развития и замедления роста ГМ после относительно нормального неонатального периода. Это Х-сцепленное заболевание, которое встречается преимущественно у женщин. Частота составляет 1:15 000-22 000 детей. Синдром Ретта вызван мутациями в гене МеСР2 на Xq28, который кодирует фактор транскрипции, связывающийся с метилированными островками CpG и глушит транскрипцию. Ребенок может нормально развиваться до 1 года, когда регрессия речевого и моторного развития и приобретенная микроцефалия становятся очевидными. Атаксическая по-ходка/мелкий кинетический тремор рук — ранние неврологические симптомы.
У большинства детей развивается своеобразное дыхание со вздохами и периодическими периодами апноэ, которые могут сопровождаться цианозом. Отличительной чертой синдрома Ретта являются повторяющиеся скручивающие стереотипные движения рук, потеря целенаправленного и спонтанного использования рук; эти признаки могут проявиться только в 2-3 года. Аутистическое поведение является типичным для всех пациентов. Генерализованные тонико-клонические судороги встречаются в большинстве случаев, но могут хорошо контролироваться противосудорожными ЛП.
Характерны расстройства питания, плохая прибавка МТ. После начального периода неврологической регрессии процесс, по-видимому, выходит на плато, с сохранением аутистического поведения. Сердечные аритмии могут привести к внезапной, неожиданной смерти с частотой, превышающей общую популяцию. Как правило, женщины доживают до зрелого возраста.
Посмертные исследования показывают значительное снижение массы ГМ (на 60-80% от нормы) при уменьшении количества синапсов, связанном с уменьшением длины и ветвления дендритов. Хотя немногие мужчины выживают с классическим фенотипом синдрома Ретта, генотипирование мальчиков без классического фенотипа, но с умственной отсталостью и др. атипичными неврологическими особенностями выявило значительное число мутаций в МеСР2. Мутации в МеСР2 выявлены у здоровых женщин-носителей, женщин с синдромом Ангельмана (Angelman) и мужчин с фатальной энцефалопатией, синдромом Клайнфельтера (Klinefelter) (47.ХХУ) и семейными Х-связанными когнитивными нарушениями. У мужчин может наблюдаться реттоподобный синдром при дупликации МеСР2.
Некоторые женщины имеют атипичный фенотип Ретта, сопровождающийся тяжелыми миоклоническими приступами в младенчестве, замедлением роста головы и остановкой развития, а также имеют мутации в др. Х-сцепленном гене, кодирующем циклин-зависимую киназу 5, которая может взаимодействовать с МеСР2 и др. белками, регулирующими экспрессию генов.
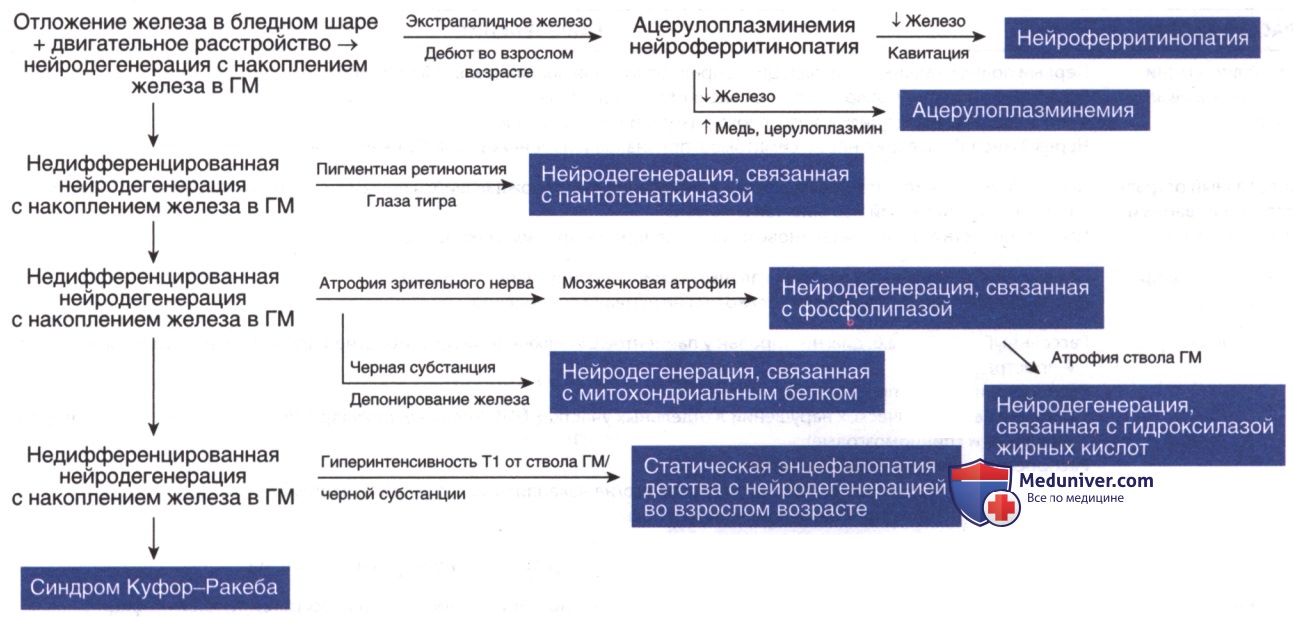
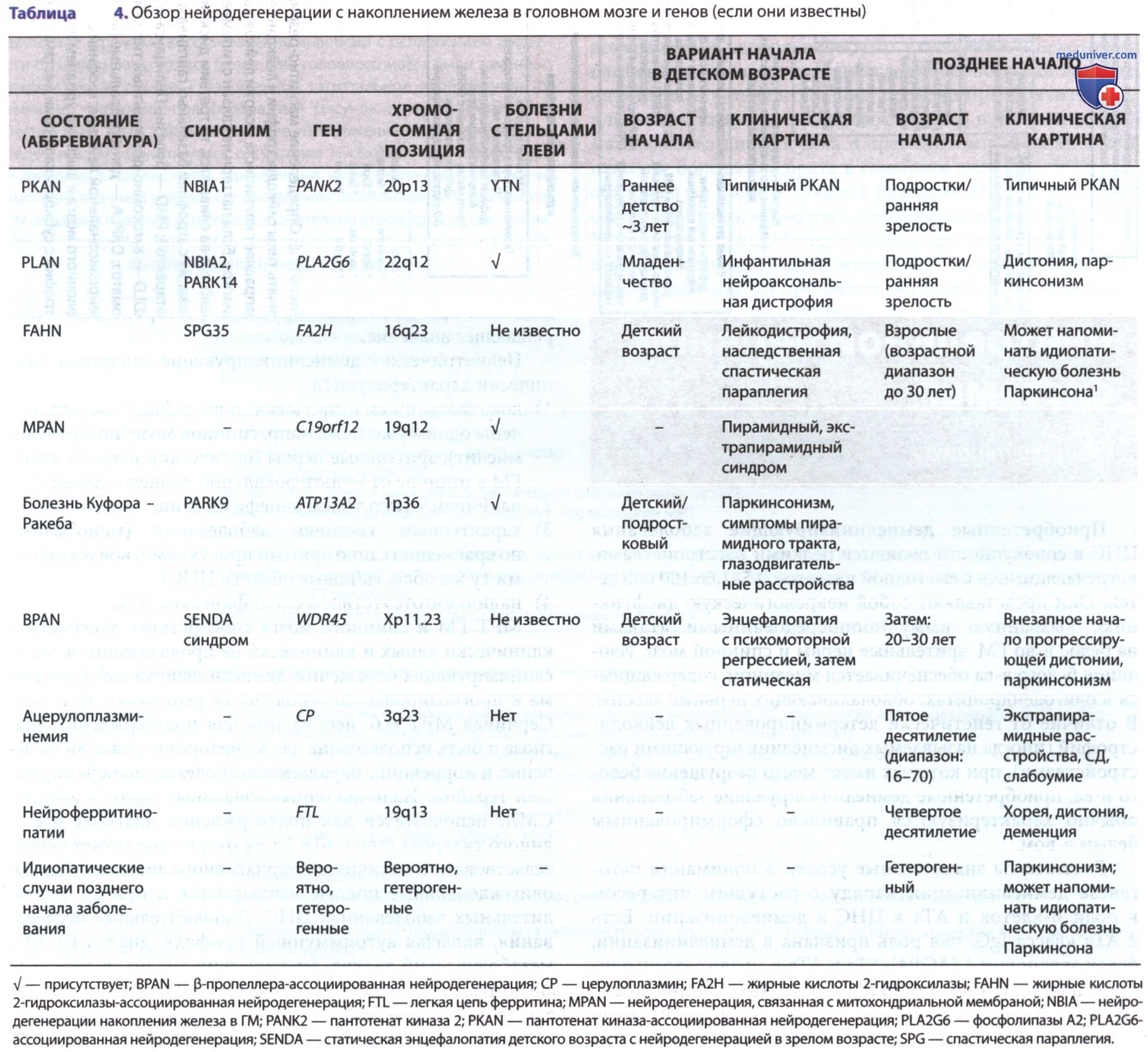
ж) Нейродегенерация с накоплением железа в мозге. Нейродегенерация с накоплением железа в ГМ представляет собой множественные возраст-зависимые расстройства, характеризующиеся экстрапирамидными симптомами, снижением и регрессией интеллекта, с отложением железа в базальных ганглиях. Существует значительная фенотипическая вариабельность этих нарушений, однако характерные изменения на МРТ показывают симметричную однородную гипоинтенсивность Т2-сигнала. Общая нейродегенерация с накопления железа в ГМ отмечена в табл. 4, а подход к их диагностике см. на рис. 4. Клинические признаки, которые сильно варьируют, могут включать дистонию, паркинсонизм, атаксию, спастичность, психические симптомы и интеллектуальные нарушения.
Рисунок 4. Клинико-рентгенологический подход к диагностике нейродегенерации с накоплением железа в головном мозге.
Лечение должно быть сосредоточено на конкретном расстройстве и обычно является симптоматическим, а не патогенетическим. Хелатирование железа не приносит большой долгосрочной пользы.