Последние достижения в области молекулярной биологии привели к лучшему пониманию этиологии многих опухолей основания черепа. Понимание клеточных механизмов, участвующих в онкогенезе, направлено на разработку новых вариантов лечения с улучшенными результатами, снижение заболеваемости, стабилизацию роста или возможности предотвращения опухолевых заболеваний. Продолжаются исследования нескольких терапевтических агентов известных путей таргетинга.
В качестве примера последних достижений в этой области будут ярко освещены вестибулярные шванномы (ВШ). ВШ обычно возникают как спорадические односторонние опухоли внутреннего слухового прохода (IAC) и мостомозжечкового угла (СРА); однако, могут быть и двусторонними, что является отличительным признаком наследственного генетического заболевания нейрофиброматоза 2 (НФ2) [Online Mendelian inheritance in Man (OMIM) #101000].
Синдром нейрофиброматоза 2 типа (НФ2) — это аутосомно-доминантное проявление биаллельной мутации 22-й человеческой хромосомы. Нейрофиброматоз 2 типа (НФ2) синдром порождает ВШ, а также менингиому, эпендимому и пресенильную катаракту.
Фундаментальное понимание молекулярных событий, ведущих к образованию опухолей, началось после идентификации гена-супрессора опухоли нейрофиброматоза типа 2 (НФ2 ген) и были подтверждения мутации в вестибулярной шванноме (ВШ) или зародышевой линии. Клинические проявления вестибулярной шванномы (ВШ) и синдрома НФ2 связаны с изменениями в НФ2 гене. Описаны различного рода мутации, влияющие на клинику НФ2, и все более понятными становятся сигнальные пути, затронутые продуктом НФ2 гена, — белком мерянном.
Для статей на сайте и другой литературы принят следующий список сокращений: «НФ2» используется для обозначения заболевания нейрофиброматоз типа 2; курсивом «НФ2» обозначен человек с геном нейрофиброматоза типа 2; и «Нф2» (Nf2) соответствует гомологу или аналогу гена экспрессии у грызунов. Мышиный Нф2 ген и человеческий НФ2 ген более чем в 98% гомологичны по аминокислотам, что делает мышь наиболее подходящей моделью для изучения. Интересно, что куры в 94% и дрозофилы в 50% также гомологичны человеческим генам по аминокислотам.
По номенклатуре nf+/+ — это дикий тип или нормальный nf ген (значение nf-/- отсутствуют обе аллели (гомозигота) и значение nf+/- — гетерозигота или только одна аллель перестает функционировать).
Вестибулярные шванномы (ВШ) могут быть разделены на односторонние спорадические солидные вестибулярные шванномы (ВШ) или двусторонние НФ2-ассоциированные шванномы. Вестибулярная шваннома (ВШ) с истинными кистами или кистозные шванномы представляют собой уникальный агрессивный фенотип, который отличает его от более доброкачественных солидных односторонних спорадических шванном. Мутации в НФ2 гене могут совпадать во всех трех типах опухоли, но уже на молекулярном уровне эти опухоли начинают отличаться от других типов.
Шванномы обычно представлены как одиночные опухоли и составляют 95% всех вестибулярных шванном (ВШ). Спорадические односторонние ВШ выявляются с частотой примерно 10-20 человек на миллион в год. Однако истинная заболеваемость может быть выше, как подчеркнули Anderson и соавт., которые продемонстрировали семь случаев шванном на 10000 исследований МРТ головного мозга (0,07%) или 70 на миллион. Спорадические опухоли, как правило, возникают в 4-ом и 5-ом десятилетии жизни, средний возраст около 50 лет. Несмотря на гистологическую доброкачественность, шванномы могут сдавливать ствол головного мозга, приводя к гидроцефалии, образованию грыжи или смерти.
Однако чаще они ассоциированы с потерей слуха, шумом в ушах, нарушением равновесия, параличом лицевого нерва и лицевыми парестезиями.
Нейрофиброматоз 2 типа (НФ2) [OMIM#101000] — аутосомно-доминантное заболевание с высокой пенетрантностью. НФ2-ассоциированные опухоли составляют около 5% всех вестибулярных шванном (ВШ). У пациентов, унаследовавших ненормальную копию НФ2 гена-супрессора опухоли, вероятность развития двусторонней ВШ 95%. Однако около половины пациентов не имеют семейного анамнеза НФ2, и, таким образом, они как основатели представляют новые зародышевые мутации, а не унаследованные. К другим НФ2 заболеваниям относятся внутричерепные менингиомы, эпендимомы, спинальные шванномы и пре-сенильная катаракта. Симптомы появляются, как правило, в 11-30 лет, но у некоторых пациентов симптомы могут присутствовать в 5-ом или 6-ом десятилетии.
Уровень заболеваемости составляет 1:33000 и до 1:40000. НФ2 не следует путать с нейрофиброматозом типа 1 (НФ1) или заболеванием фон Реклингхаузена [OMIM #162200]. НФ1, сопровождающийся множественными периферическими нейрофибромами, вызывается мутацией в НФ1 гене-супрессоре опухоли 17 хромосомы.
Спектр фенотипической экспрессии в НФ2 колеблется в широких пределах. В прошлом более сильный клинический фенотип назывался типом Уишарта. Кроме того, пациенты с двусторонними ВШ часто страдают спинномозговыми опухолями, для которых характерна манифестация симптомов в позднем подростковом возрасте или в начале второго десятилетия. Менее тяжелые фенотипы были названы типом Гарднера, они отличаются поздним началом двусторонних шванном и реже ассоциированы с внутричерепными или спинномозговыми опухолями. В действительности, спектр заболеваний не всегда позволяет проводить четкое различие, скорее следует придерживаться совокупности признаков.
В основе другой классификации нейрофиброматоза 2 типа (НФ2) лежат сегментарные или мозаичные категории НФ2. Причиной сегментарной НФ2 может являться соматический мозаицизм, в этом случае мутация происходит не в ДНК зародышевой линии, а позже в эмбриогенезе; таким образом, только часть клеток пациентов несет мутации, и болезнь проявляется в ограниченных областях. Напротив, больные с семейной НФ2 наследуют одну мутацию от родителя в момент зачатия и все клетки несут один мутантный аллель. Kluwe и соавт. предполагают, что мозаицизм может составлять 25% случаев любого подтипа НФ2 среди пациентов, чьи родители не имеют проявлений заболевания. Проявления двусторонних вестибулярных шванном (ВШ) встречается у пациентов с соматическим мозаицизмом, если произошла постзиготная мутация на ранней стадии эмбриогенеза.
Однако, если постзиготная мутация произошла позже в развитии, то характерны атипичные проявления, или сегментарный нейрофиброматоз 2 типа (НФ2), при этом у пациента односторонняя вестибулярная шваннома, а с противоположной стороны — дополнительная внутричерепная опухоль, такая как менингиома. Риск передачи нейрофиброматоза 2 типа (НФ2), вызванного мозаицизмом, для будущего поколения, в отличие от традиционных форм нейрофиброматоза 2 типа (НФ2), низкий.
Кистозные вестибулярные шванномы (ВШ) — особо агрессивная группа односторонних, спорадических шванном, которые вторгаются в ближайшие черепные нервы и смещают их по всей протяженности опухоли. Кистозные вестибулярные шванномы (ВШ) ассоциированы с различными внутриопухолевыми или внеопухолевыми кистами. Кроме того, в кистозных опухолях выявлена более высокая степень атипии ядер. Следует тщательно проводить различие между истинными шванномами и очень распространенными гетерогенными шванномами, которые не столь агрессивны в своем клиническом проявлении. На магнитно-резонансной томографии (МРТ) кистозные области опухоли гиперинтенсивны на Т2-взвешенных изображениях, при контрастировании кисты гадолинием усиления интенсивности изображения не происходит.
Гадолиний усиливает компоненты вне кист при кистозных опухолях таким же образом, как при односторонних и НФ2-связанных шванномах. Кистозные опухоли могут расти очень быстро, и трудно лечатся, вызывая высокий процент потери слуха, паралича лицевого нерва и послеоперационных рецидивов. При сравнении солидных опухолей аналогичного размера, уровень полного паралича лицевого нерва (по шкале Хаус-Бракмана класс VI) при хирургическом удалении кистозных опухолей составил 41%, при солидных односторонних шванномах 27%. При стереотаксической лучевой терапии более вероятно продолжение роста кистозных опухолей и паралич лицевого нерва, чем односторонних спонтанных или НФ2-ассоциированных шванном.
Несмотря на эффективность хирургической и лучевой терапии вестибулярной шванномы (ВШ), осложнения лечения по-прежнему остаются проблематичными. Ниже следует краткий обзор последних открытий и достижений в молекулярной биологии ВШ.
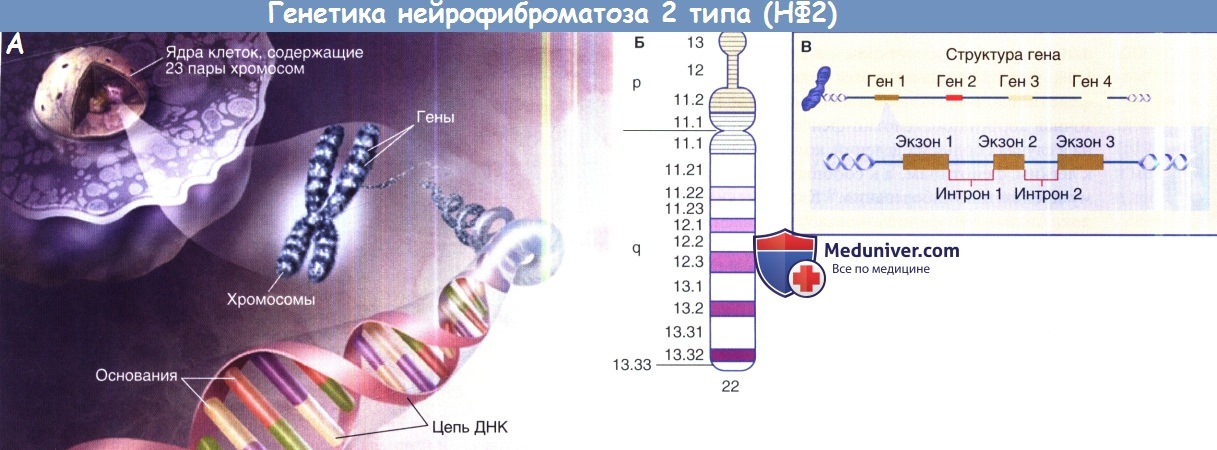
А Изображение, показывающее связь хромосом, генов оснований ДНК нуклеотидов.
Б. Схема одного плеча 22 хромосомы, изображающая р и q сегменты хромосомы. В, модель человеческого гена.
Информация, содержащаяся в генах, используется для синтеза белка, делится на экзоны (кодирующие белок) и интроны (некодирующие последовательности).
а) Ген нейрофиброматоза 2 типа (НФ2 ген). Путем генного и физического картирования НФ2 ген был локализован в 22 хромосоме, в локусе q12. Исследования позиционного клонирования привели к открытию НФ2 гена в 1993 году двумя независимыми группами, Rouleau и соавт. и Trofatter и соавт. Был выявлен ген, часто мутирующий в НФ2-связанных вестибулярной шванномой (ВШ). С того времени мутации НФ2 гена были обнаружены не только в НФ2-ассоциированных опухолях, но и в спорадических односторонних и кистозных шваномах. Помимо этого мутации НФ2 гена часто выявляются в менингиомах, а иногда и в других типах опухолей, таких как мезотелиомы.
б) Человеческие НФ2 мутации и их клиническая корреляция. Специфические НФ2 мутации в вестибулярной шванноме (ВШ) были описаны в спорадических односторонних шванномах и НФ2-связан-ных шванномах. Частота, тип и распространение НФ2 мутаций показали различия между спорадическими и семейными НФ2-опухолями. Точечные мутации составляли большинство мутаций, выявленных у НФ2 пациентов, в то время как малые удаления составляли большинство мутаций, обнаруженных в спорадических односторонних опухолях.
Две группы недавно опубликовали большие базы данных НФ2 мутаций. Ahronowitz и соавт. представили мета-анализ 1070 небольших генетических изменений, в том числе 42 внутригенных изменений одного целого экзона или больше, и 29 целых делеций гена и грубых хромосомных перестроек. В целом соматические события, выявленные путем анализа опухоли показали существенно отличающийся генетический профиль, по сравнению с конституциональными событиями, зарегистрированными в лейкоцитах больных. Более половины мутаций, обнаруженных в опухоли, были мутациями сдвига рамки считывания.
Удаление единичных нуклеотидов (А<Т<Ц< или Г) из генов действует особенно разрушительно, потому что все кодоны, расположенные ниже мутации, меняются. При сравнении конституциональных или системно наследуемых изменений выявлены, в основном, нонсенс-мутации и сплайс-сайт мутации. Соматические события также заметно отличались при менингиомах, где большинство мутаций находились в пределах 5-прайма четыре, эзрин, радиксин и моэзин (FERM) домена транскрипта с полным отсутствием мутаций в экзонах 14 и 15. Менее 10% малых изменений были неусеченными или повреждениями с неполным уничтожением белка. Эти изменения были сгруппированы во 2 и 3 экзонах, давая основания для предположения, что эта область может быть особенно важна для подавления активности опухоли в белке.
Мы и другие исследователи показали, что частый образец мутации — замена аргинин кодонов (ЦГА) тимин стоп-кодоном (ТГА) (Этот тип стоп-кодона не только завершает транскрипцию мРНК, но и зачастую приводит к деградации уже расшифрованной мРНК) и в вестибулярной шванноме (ВШ) и в менингиомах, вызывая сокращение повреждения.
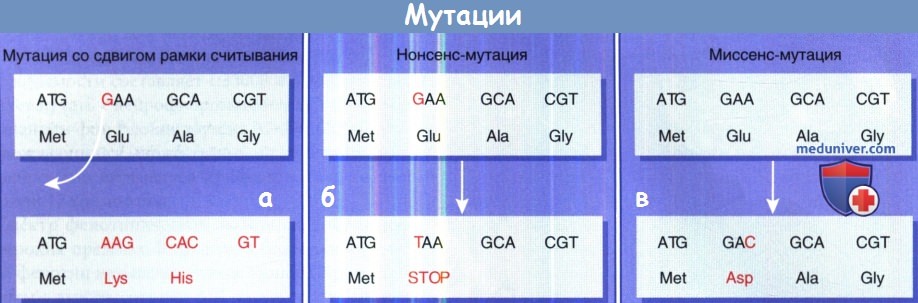
А. Делеция единичного нуклеотида (А, Т, С и G) гена особенно нарушает влияние на ген,
так как делеция одного нуклеотида изменяет все нижерасположенные кодоны, в результате происходит мутация сдвига рамки считывания.
Удаление «G» основания меняет нижерасположенное кодирование, таким образом вместо кодирования для...
Glu-Ala-Gly, мутировавших генов, теперь кодирует для... Lys-His... расположенные ниже мутации.
Б. нонсенс-мутация одного нуклеотида, который преобразует кодон, что указывает аминокислоты в прекращении кодон.
Это вызывает укорочение белка после того, как трансляция мРНК останавливается на этом новом терминирующем кодоне без коррекции при спуске вниз.
Эффект нонсенс-мутаций зависит от того, сколько белка теряется.
В. Точечная мутация или изменение одного нуклеотида способны изменить кодон так, что определяется другой белок.
Это называется миссенс-мутации, определена неверная аминокислота. Поэтому кодируемый геном белок изменяет одиночную аминокислоту.
Часто это не оказывает особого эффекта на белок, в большинстве случаев возможно несколько изменений аминокислот без влияния на их биологическую функцию.
Химические соединения, участвующие в конфигурации этих нуклеотидов, делают их менее стабильными и возникает большая вероятность спонтанных мутаций не только в НФ2, но и многих других опухолях. Не были зарегистрированы мутации в 16 и 17 экзонах, а небольшое их количество отмечено в 9 экзоне.
Исследования определения возможности влияния генотипа на прогноз тяжести заболевания показали, что делеции, вызывающие усечение НФ2 белка, имеют более тяжелый фенотип в НФ2 родословных, хотя миссенс-мутации или небольшие вставки в НФ2-кодирующей области связаны с легким фенотипом. Впрочем, встречаются исключения, и некоторые миссенс-мутации были связаны с тяжелым фенотипом. Из этого ясно, что место расположения мутации внутри гена может быть важно, как например, миссенс-мутации в а-спиральном домене белка НФ2 представляются связанными с менее тяжелым фенотипом, чем в консервативном домене ERM.
Недостаточная корреляция генотип-фенотипа также была отмечена в случаях больших делеций, которые могли бы привести к легким фенотипам, а к сообщавшемуся ранее тяжелому проявлению заболевания. Клинические исследования показывают, что фенотипическое проявление имеет более тесную взаимосвязь в семьях, чем между семьями, но даже внутри семьи существует изменчивость. Статистический анализ возраста начала НФ2, начала снижения слуха и количества внутричерепных менингиом, показал существенные внутрисемейные корреляции. Неизвестно, происходит ли эта изменчивость случайно, как эпигенетическое явление, или путем модификации генов в других локусах.
Эпигенетические явления вызываются не изменением в кодирующей последовательности ДНК, а скорее изменением экспрессии генов во время развития или клеточной пролиферации. В качестве примера можно привести метилирование сегментов промоторной области гена НФ2, подавляющее экспрессию гена. Регион НФ2-кодирования включает 17 экзонов, охватывающих 90 тысяч пар оснований (кВр) ДНК 22 хромосомы. Zucman-Rocci и соавт. путем обширного скрининга всего гена НФ2 выявили частоту мутаций в ВШ, последняя составила 84%. Таким образом, у части пациентов могут существовать дополнительные механизмы инактивации НФ2 гена. Мутация или метилирование в регулирующей области НФ2 гена были предложены в качестве возможного механизма инактивации гена.
Посттранскрипционный альтернативный сплайсинг и дифференциальное полиаденилирование могут также рассматриваться в качестве возможных методов инактивации НФ2 гена. Полиаденилирование указывает на переменное число аденинов, которые крепятся на 3‘ конце мРНК, что может играть роль в пути регулирования гена. Альтернативный сплайсинг относится к различным образцам сращивания экзонов, который возникает в этом случае НФ2 гене, в результате чего образуются различные изоформы гена. Две наиболее распространенные изоформы НФ2 имеют 16 экзон или 17 экзон вне сплайсинга.
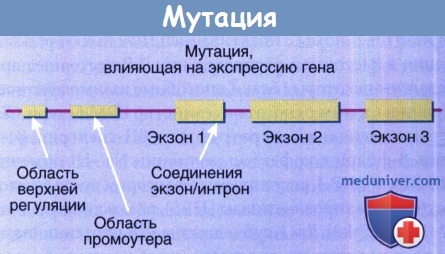
Мутации могут возникать выше самого гена в промоутере или областях регуляции или на границе между нитронами и экзонами (места соединения).
Мутации в промоутерных или регуляторных регионах могут привести к чрезмерной или недостаточной экспрессии гена,
в то время как мутации на границе интрона/экзона могут вызывать аберрантный сплайсинг.
Аберрантный сплайсинг способен удалить часть получаемого белка, добавить новую секцию аминокислот или привести к сдвигу рамки считывания.