За исключением генов, локализованных на половых хромосомах (X и Y), мы наследуем по две копии каждого гена. В большинстве случаев активны и материнские, и отцовские копии генов. Поэтому при наследовании заболевания передающегося аутосомно-доминантным путем обычно неважно, кто из родителей передал заболевшему ребенку ген с однонаправленной мутацией.
Однако существует множество генов, у которых активна или функциональна только лишь материнская или отцовская копия, а вторая неактивна, поэтому ее называют импринтинговой. Импринтинговые гены обычно вовлечены в период эмбрионального роста и поведенческого развития, но время от времени они могут функционировать несоответствующим образом, как онкогены или как ген подавляющий опухоль.
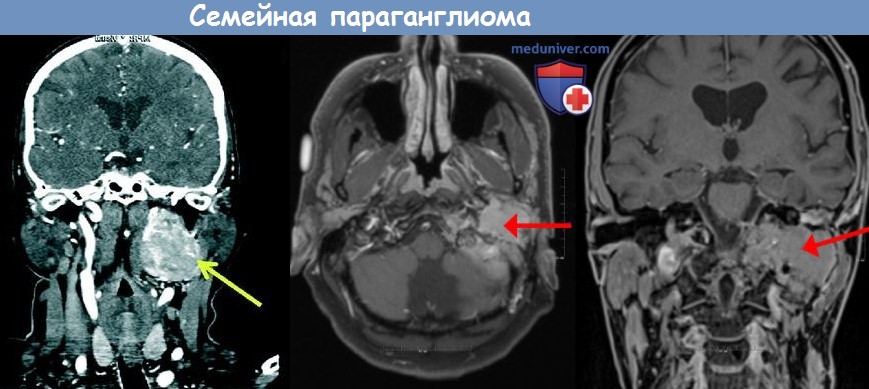
Семейные параганглиомы (ПГ) наследуются по аутосомно-доминантному типу с материнским импринтингом. Вследствие этого, когда ребенок наследует ген параганглиомы (ПГ) от матери (независимо от того, страдает она сама или нет), он не подвергается воздействию, являясь лишь скрытым носителем мутированного гена. С другой стороны, наследуя ген параганглиомы (ПГ) от отца, ребенок будет иметь ПГ вне зависимости от того, страдает этим заболеванием отец или нет.
Впоследствии, страдающий или нестрадающий ребенок, унаследовавший ненормальный ген параганглиомы (ПГ) может передать этот ген сыну/дочери; он/она будут иметь детей, страдающих этим недугом только в том случае, если унаследуют заболевание от отца. Такая необычная форма неполной генетической пенетрантности определяется специфической для пола генной модификацией в период гаметогенеза.
Принимая во внимание описанные выше необычные модели наследования, трудно определить, в каких случаях «спорадические» параганглиомы (ПГ) таковыми и являются, а в каких имеет место замаскированное под спорадический вариант наследование. По всей вероятности, многие спорадические опухоли являются наследственными, и встречаемость наследственных опухолей в значительной мере выше чем 1 на 10 параганглиом (ПГ).
Схожие по их наследственному прототипу спорадические опухоли также демонстрируют потерю гетерозиготы PGL1 и PGL2. Bikhazi и соавт. продемонстрировали потерю гетерозиготы в 38% случаев спорадических случаев опухолей каротидного гломуса и гломусных опухолей при тестировании маркеров, расположенных на PGL1 и PGL2. Это косвенное подтверждение того, что треть спорадических опухолей в действительности может относиться к наследственным.
Так же, как и нейрофиброматоз 2 типа (НФ2), спорадический и наследственный ПГ видимо имеют общую генетическую этиологию.
Поиски генов, ответственных за параганглиомы (ПГ) в большой степени были достигнуты благодаря разработкам по человеческому геному и наличию большого количества семей с наследственной параганглиомой (ПГ).
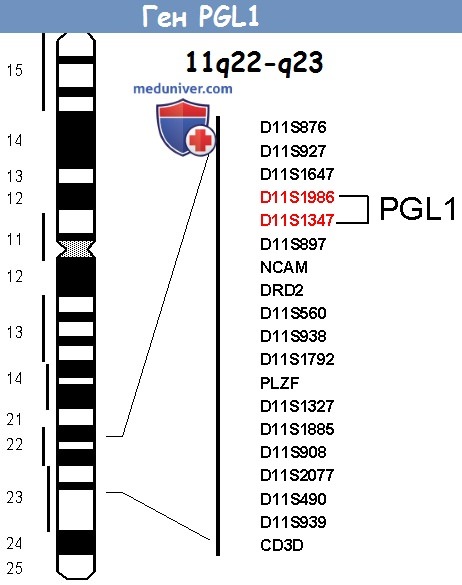
а) Первый локус: PGL1. Генетический анализ сцепления большой голландской семьи с наследственными ПГ обнаружил ген, названный PGL1, на коротком плече 11 хромосомы, 11q22-q23. Эти данные были подтверждены при исследовании семей в Северной Америке, где локализация также находилась на 11q23.
Опухолевые клетки, взятые от больных с PGL1 мутацией, показали избирательную потерю информационной ДНК 1lq хромосомы, содержащей ген PGL1, обеспечивающую генетическую поддержку для генетического импринтинга. Явление, когда ДНК удаляется, называется потерей гетерозиготности. Такая потеря, связанная с формированием опухоли, четко указывает, что ген PGL1 является подавляющим опухоль.
Согласно гипотезе опухолевой супрессии развитие опухоли происходит при утрате обеих копий подавляющего опухоль гена, а для предотвращения развития новообразования достаточно единственной функционирующей копии. Гены НФ2 и ретинобластомы являются примерами опухолевых супрессорных генов. Потеря функции одного из супрессорных генов при наследовании мутированной аллели и последующая случайная мутация второй аллели приводят к онкогенезу.
Этот феномен, описанный Knudson, известен как гипотеза «двойного попадания». При семейной параганглиоме (ПГ), как при нейрофиброматозе 2 типа (НФ2), наследуемые в первую очередь «попадания» предрасполагают к развитию мультицентрических опухолей вследствие множественных случайных вторичных «попаданий».
Ген, ответственный за PGL1, был клонирован в пределах большой хромосомной последовательности 11q23 при скрининге генов-кандидатов, участвующих в кислородном обмене этой области. Данный ген, известный как SDHD (для субъединицы D сукцината-убихинона оксиредуктазы) кодирует маленькую субъединицу митохондриального цитохрома b (cybS), вовлеченную в аэробный метаболизм цикла Кребса.
При наблюдении пяти семей с наследственной параганглиомой (ПГ) были идентифицированы мутации единичной пары оснований, приводящие к выпадению функции гена SDHD. Нонсенс-мутация была выявлена в двух семьях; оказалось, что она лежит в основе раннего отсекания при формировании белка SDHD и последующей утрате образования cybS.
На примере других трех семей была показана частота миссенс-мутации с изменением единственной аминокислоты, предположительно влияющей на кардинальное изменение конформации cybS с отсутствием функционирования. Интересно, что не был найден типичный постгаметогенез материнского импринтинга, но была обнаружена биаллельная экспрессия и материнского, и отцовского генов в тканях организма.
Это указывает на то, что случае, когда нормальная материнская аллель позднее импринтируется или утрачивается, мутировавшая отцовская аллель, кодирующая мутировавший SDHD, ведет к развитию опухоли с утратой активности подавляющей опухоль. Исследование этого гена при скрининге членов семей с риском наследственного ПГ, несомненно даст более точные заключения.
б) Второй локус: PGL2. Исследование другой большой неродственной голландской семьи с наследственным ПГ обнаружило второй локус, PGL2, на коротком плече 11 хромосомы. Этот локус хромосомы 11q13 скрывал другой ген ПГ, который, как выяснилось при генетическом исследовании, абсолютно отличался от PGL1.
в) Третий и четвертый неимпринтинговые локусы: PGL3 и PGL4. На третий локус, PGL3, была обследована большая немецкая семья с наследственной параганглиомой (ПГ) Маркеры, располагающиеся на локусах 11q23 и 11q13, ассоциированные с PGL1 и PGL2, исключали наличия сцепления с этой областью. Наследование в этой семье также было по аутосомно-доминантному типу, как и при PGL1 и PGL2, но отсутствовал материнский импринтинг. В этой семье ген локализовался на хромосоме 1q21.
Так как мутации в SDHD были расценены как главная причина параганглиомы (ПГ) 1 типа, то Niemann и Muller исследовали SDHC и в этой семье. Ими было обнаружено замещение гуанина на аденозин в 1 экзоне SDHC у всех членов семьи, которые страдали ПГ, и не было обнаружено ни у одного не страдающего ПГ члена семьи. Мутация разрушила инициирующий кодон гена ATG в последовательности нуклеотидов 958.
В митохондриальном комплексе II, SDHA и SDHB составили каталитические домены и закрепились во внутренней цитоплазматической оболочке как субъединицы SDHC и SDHD. Скрининг при наличии изменений SDHB помог обнаружить мутации, ассоциированные с PGL4; мутации в SDHB также ассоциированы со злокачественной феохромоцитомой. PGL3 и PGL4 не зависят от материнского импринтинга.
г) Генетический скрининг. Быстрое продвижение молекулярной генетики и биологии позволило практикующим оториноларинлогогам проникнуть в таинство этиологии редкой, но при этом хорошо описанной опухоли головы и шеи. При исследовании пациента с ПГ должен быть собран расширенный семейный анамнез, что поможет обнаружить и других членов семьи с клиническими проявлениями опухолей головы и шеи и подозрением на ПГ.
При подозрении на наследственную параганглиому (ПГ) должна быть составлена генеалогическая схема, и все члены семьи должны пройти генетическое исследование. При достаточно большой родословной может быть проведен генетический анализ и определен семейный ПГ, от PGL1 до PGL2. На основании генетических данных выявляются лица со скрытой дефектной хромосомой, тем самым и риском развития опухоли. Члены семьи с передаваемой отцовской аллелью, подверженные риску формирования параганглиомы (ПГ), должны динамически наблюдаться и исследоваться как клинически, так и рентгенологически, а также проходить генетическое консультирование.
Тех, кто наследует дефектный ген по материнской линии, относят к клинически скрытым носителям и им также необходимо генетическое консультирование. Пациентам, не считающимися носителями, можно сообщить, что у них нет повышенного риска образования опухоли в пределах общей популяции.
В качестве альтернативного варианта полученная из крови пациента ДНК может быть протестирована на мутацию и SDHD, используя полимеразную цепную реакцию и прямое секвенирование ДНК. Если мутация обнаружена, то члены семьи должны быть протестированы для того, чтобы понять, имеют ли они скрытые мутации и вследствие этого риск формирования опухоли. Генетический скрининг позволяет надеяться на идентификацию опухоли на ранних стадиях, что позволит снизить частоту заболеваемости и смертности.
В конце концов, идентификация генетической перестройки, встречающаяся и при наследственном, и при спорадическом ПГ, должна привести к возможности воздействия на рост опухоли на генетическом уровне.
Базовое понимание генетики принципиально важно для отологов и нейроотологов. Генетический скрининг и генетическое тестирование постепенно становятся частью повседневной практики. В ближайшем будущем генная терапия кардинально изменит подход к лечению глухоты и опухолей височной кости и мостомозжечкового угла.
Параганглиомы головы и шеи — редкие и в основном доброкачественные опухоли. Синдромы наследственных параганглиом представлены аутосомно-доминантными состояниями с повышенным риском мультифокальных опухолей симпатической и парасимпатической нейроэндокринной системы. Примерно 30% параганглиом у пациентов представлены мутациями зародышевой линии, мутации несут все клетки организма (в отличие от соматических мутаций, которые можно найти только в опухолевых тканях). Более трети этих пациентов не имеют предшествующей семейной истории, тем самым начиная новые мутации.
От 10 до 15% параганглиом вызваны мутациями в генах сукцинатдегидрогеназы (SDH) или их якорными субъединицами В, С, или D. Они могут являться гормонами секреции. Злокачественные параганглиомы были, в частности, связаны с мутациями в SDHB, но были также отмечены и в SDHD. Гены сукцинатдегидрогеназы ответственны за белок, являющийся малой частью комплекса цитохрома b в митохондриальном комплексе II, одном из пяти агрегатов или комплексов белков, которые образуют цепь переноса электронов. При производстве реактивных молекул кислорода этот путь считается важным в сигнализации и обнаружении напряжения кислорода. Ген, кодирующий SDHD, состоит из четырех экзонов и дает 159 аминокислотных полипептидов.
Другие мутации параганглиом оказались связаны с белками домена пролилгидроксилазы (PHD), играющими важную роль в регулировании индуцируемого гипоксией фактора (HIF), который индуцирует экспрессию генов, участвующих в ангиогенезе, эритропоэзе и клеточном метаболизме, пролиферации и выживании. Мутации в домене 2 гена пролилгидроксилазы (PHD2) недавно были описаны у пациента с эритроцитозом и рецидивирующими параганглиомами.