Пигментный ретинит (retinitis pigmentosa, RP) — термин, используемый для обозначения генетически гетерогенной группы заболеваний, проявляющихся на ранних стадиях никталопией и сужением поля зрения. Электроретинограмма или не регистрируется, или же выявляется дисфункция фоторецепторов с более тяжелым поражением палочек, чем колбочек. Дисфункция только системы палочек не характерна, но может наблюдаться на ранних стадиях заболевания. Болезнь часто дебютирует в детстве и может наследоваться по аутосомно-рецессивному, аутосомно-доминантному и Х-сцепленному механизму. Изменения могут быть ограничены тканями глаза или же сопровождаться более обширной системной патологией.
а) Симптомы и клиника пигментного ретинита. У детей с пигментным ретинитом заболевание может манифестировать ночной слепотой, симптомами, связанными с обширными дефектами поля зрения или поражением центральных отделов сетчатки. Возраст дебюта крайне вариабелен. У некоторых пациентов при отсутствии жалоб на никталопию заболевание выявляется во время плановой офтальмоскопии. При наличии пигментного ретинита у близкого родственника ребенок в раннем возрасте направляется на обследование для исключения патологических изменений.
В большинстве случаев острота зрения изначально нормальная; позже ухудшение зрения может развиваться вследствие задней субкапсулярной катаракты, макулярного отека или поражения макулы. На ранних стадиях заболевания развиваются изменения поля зрения в виде мелких скотом на средней периферии, обычно в верхних квадрантах. Постепенно они сливаются и образуют классическую периферическую кольцевую скотому. Со временем поля зрения все больше сужаются, часто сохраняется лишь островок поля зрения на крайней периферии в височном квадранте.
При секторальном пигментном ретините, обычно поражающем нижне-носовой квадрант, двусторонний верхне-височный дефект поля зрения может стать поводом для излишнего обследования с целью исключения поражения хиазмы — но граница височного дефекта поля зрения при пигментном ретините не проходит по средней линии.
Изменения глазного дна на ранних стадиях пигментного ретинита вариабельны и у маленьких детей не выражены. Самыми ранними симптомами могут быть легкие изменения пигментного эпителия на средней периферии, часто сопровождающиеся появлением мелких белых точек на уровне ПЭС. Позже отложения пигмента выявляются в экваториальной сетчатке, могут отмечаться сужение артерий и бледность диска зрительного нерва. У некоторых детей аномальная пигментация может отсутствовать (беспигментный ретинит, RP, sine pigmento) и, еще реже, могут выявляться множественные белые отложения по всей поверхности сетчатки (белоточечный ретинит — retinitis punctata albescens).
На развитой стадии заболевания наблюдается классическая картина глазного дна: бледность диска зрительного нерва, сужение артериол сетчатки, периферическая атрофия пигментного эпителия и интраретинальная пигментация по типу «костных телец». Возможно развитие и других изменений, в том числе появление клеточных элементов в стекловидном теле, задней субкапсулярной катаракты, друз диска зрительного нерва и макулярного отека. Иногда наблюдаются ретиноваскулярные изменения, аналогичные изменениям при болезни Коутса.
Фенотипически картина изменений сетчатки одинакова при большинстве форм пигментного ретинита, хотя тяжесть заболевания у различных индивидов может варьировать в широких пределах. Однако существует несколько ген-специфичных фенотипов. Аутосомно-доминантный секторальный пигментный ретинит обычно связан с мутациями гена родопсина, а нечасто встречающаяся аутосомно-рецессивная форма пигментного ретинита (RP12) с сохранением параартериолярного ПЭС связана с мутациями CRB1. Две рано дебютирующих формы аутосомно-рецессивного пигментного ретинита, вызываемые мутациями RLBP1, сопровождаются появлением множества белых отложений на уровне ПЭС (ньюфаундлендская палочко-колбочковая дистрофия и ботническая дистрофия).
Пигментный ретинит.
Атрофия пигментного эпителия на средней периферии с мелкими белыми точками на уровне ПЭС.
б) Электрофизиология и психофизика. На электроретинограмме при пигментном ретините выявляется паттерн, отражающий палочко-колбочковую дисфункцию, при этом палочковые ответы изменены сильнее, чем колбочковые. Отмечается значительная вариабельность тяжести заболевания, частично зависящая от природы мутации, механизма наследования и возраста пациента. На поздних стадиях пигментного ретинита электроретинограмма может не регистрироваться, или же фиксируются слабые остаточные колбочковые ответы. При легком течении или на ранних стадиях заболевания наблюдается уменьшение амплитуды палочковых а- и b-волн, может увеличиваться зависящая от функции палочек латентность b-волны. Ответы на ритмическую стимуляцию 30 Гц обычно замедлены и ослаблены, что указывает на генерализованную дисфункцию системы колбочек.
Электроокулография (electro-ocuologram, EOG) ненамного информативнее, чем ЭРГ, но ее выполнение у маленьких детей затруднено. Функция макулы может быть сохранена, и при исследовании функции центральных отделов сетчатки, например, при паттерн-ЭРГ или мультифокальной ЭРГ, могут регистрироваться минимальные нарушения, несмотря на почти не регистрируемую ганцфельд-ЭРГ. Паттерн-ЭРГ также информативна при диагностике поражения центральной зоны сетчатки до развития видимых изменений.
в) Секторальный пигментный ретинит. Термином секторальный пигментный ретинит следует обозначать те формы пигментного ретинита (обычно аутосомно-доминантные), при которых результаты функциональных исследований указывают на частичную утрату функции; пигментные аномалии обычно ограничены нижними отделами сетчатки. Аналогичное секторальное поражение может иногда наблюдаться у женщин-носителей Х-сцепленного пигментного ретинита или на ранних стадиях генерализованного пигментного ретинита. На ЭРГ при истинном секторальном пигментном ретините обычно регистрируется снижение амплитуды, но изменения латентности отсутствуют; значительное увеличение латентности указывает на генерализованную дисфункцию, даже при наличии ограниченных пигментных изменений.
В случае изолированного заболевания важное значение имеет обследование других членов семьи, поскольку заболевание часто протекает бессимптомно. Доминантный секторальный пигментный ретинит часто связан с мутациями гена родопсина. У детей секторальный пигментный ретинит обычно протекает бессимптомно и диагностируется при плановой офтальмоскопии или при обследовании всей семьи.
Дефекты поля зрения при выполнении периметрии по Гольдману соответствуют сектору клинически пораженной сетчатки. Однако при периметрии в условиях темновой адаптации может выявляться повышение порогов чувствительности палочек и колбочек внешне не измененной сетчатки, что свидетельствует о дисфункции фоторецепторов вне клинически измененной зоны. Темновая адаптация палочек может быть крайне замедленной. ЭРГ обычно относительно сохранна, отмечается легкое или умеренное снижение амплитуды ответов палочек и колбочек. Заболевание прогрессирует медленно и поражение обычно ограничено клинически измененным сектором. Прогноз для зрения хороший.
г) Аутосомно-доминантный пигментный ретинит с неполной пенетрантностью. Вариабельность проявлений — обычное явление при аутосомно-доминантном пигментном ретините, но истинная неполная пенетрантность встречается нечасто. Однако в некоторых семьях, в которых отмечается ранний дебют и тяжелое течение заболевания, неполная пенетрантность выражена и встречается часто. При аутосомно-доминантном пигментном ретините с неполной пенетрантностью отмечается генетическая гетерогенность, идентифицированы мутации нескольких генов, в том числе RP9 и PRPF31.
Генетическое консультирование семей, в которых наблюдается неполная пенетрантность, может быть проблематичным из-за часто встречающихся бессимптомных носителей генов, но в некоторых случаях возможно проведение современной генетической диагностики.
д) Х-сцепленный пигментный ретинит. Х-сцепленный пигментный ретинит—тяжелое прогрессирующее заболевание, дебютирующее никталопией в возрасте около десяти лет и приводящее к слепоте в третьем-четвертом десятилетии жизни. У пациентов часто развивается миопия и рано выявляются аномалии глазного дна и изменения на ЭРГ. При наличии отягощенного семейного анамнеза или больного родственника мужского пола диагноз не вызывает затруднений. Если семейный анамнез не отягощен, информативно обследование близких родственников женского пола, поскольку диагностика носительства Х-сцепленных генов помогает подтвердить диагноз.
Состояние носительства Х-сцепленного пигментного ретинита точно диагностируется у женщин при наличии больного родственника — сына или отца, женщина, таким образом, является облигатным носителем гена, или же носительство патогенной мутации можно подтвердить с помощью молекулярного генетического анализа. У других родственников женского пола диагностика носительства может быть проведена на основании изменений глазного дна гетерозигот или результатов электрофизиологического или психофизического обследования.
Аномалии глазного дна часто встречаются у гетерозигот по Х-сцепленным генам: может определяться выраженный «ковровый» («tapetal») рефлекс заднего полюса или легкое истончение пигментного эпителия в экваториальной зоне и пигментация. У гетерозигот обычно регистрируется аномальная и асимметричная электроретинограмма. Наиболее часто регистрируемая аномалия, вероятно, это задержка ответа на стимуляцию мельканием 30 Гц, но также встречается снижение амплитуды палочкового ответа. В большинстве случаев носительство Х-сцепленных генов может быть диагностировано с помощью комбинации офтальмоскопии и электрофизиологических методов. Во многих семьях возможно проведение молекулярной генетической диагностики, и этот метод все шире используется для идентификации гетерозигот.
У некоторых носителей Х-сцепленных мутаций дисфункция сетчатки может прогрессировать, носители старшего возраста могут предъявлять жалобы на никталопию, у них могут выявляться дефекты поля зрения и более обширная пигментация сетчатки. У некоторых женщин-носителей может развиваться тяжелое поражение в относительно молодом возрасте; однако наблюдается выраженная межсемейная и внутрисемейная вариабельность. Тяжелые поражения женщин могут развиваться при нарушении инактивации Х-хромосомы, вследствие чего мутантная X-хромосома, содержащая мутантный аллель, остается активной в большинстве клеток.
е) Односторонний пигментный ретинит. Описаны односторонние изменения глазного дна, но зафиксирован только один случай одностороннего пигментного ретинита, возникший в большой семье с пигментным ретинитом. Пациент из этой семьи с односторонним пигментным ретинитом, являлся носителем часто встречающейся нонсенс-мутации гена RPU частой причины аутосомно-доминантного пигментного ретинита. Таким образом, вероятно, соматическая мутация возникла в клетке-предшественнике в процессе развития нормальной ткани сетчатки, вследствие чего реализовался эффект мутации гена RP1, Большинство остальных случаев, по всей вероятности, не подтверждались генетически и на самом деле являлись поствоспалительными, посттравматическими изменениями или последствиями ишемии сетчатки.
У некоторых пациентов при манифестации заболевания наблюдаются асимметричные изменения глазного дна, и один глаз клинически кажется не пораженным, но с помощью психофизических исследований и электроретинографии выявляются двусторонние нарушения функции.
Секторальный пигментный ретинит.
Пигментные изменения сетчатки ограничены нижними квадрантами.
ж) Дифференциальный диагноз пигментного ретинита. Другие заболевания могут ошибочно приниматься за пигментный ретинит из-за развития никталопии или из-за сходной картины глазного дна. Другие наследственные дистрофии обычно можно дифференцировать от пигментного ретинита по клиническим или электрофизиологическим данным. Хотя данные анамнеза и осмотра позволяют исключить многие приобретенные причины пигментной ретинопатии, вероятно, что в некоторых случаях за спорадический пигментный ретинит принимаются приобретенные заболевания сетчатки или пигментного эпителия.
з) Генетика пигментного ретинита. Пигментный ретинит может наследоваться по аутосомно-доминантному (АD), аутосомно-рецессивному (AR) или Х-сцепленному (XL) рецессивному механизму. В пределах этих подтипов наблюдается генетическая гетерогенность. Относительная встречаемость различных типов наследования в различных сериях наблюдений варьирует в широких пределах, но у 50% пациентов семейный анамнез не отягощен по пигментному ретиниту или по близкородственным бракам. Маловероятно, чтобы у всех этих пациентов была аутосомно-рецессивная форма заболевания. У некоторых мужчин это может быть Х-сцепленная форма, передавшаяся от женщин — бессимптомных носителей; другие случаи могут быть вызваны вновь возникшими аутосомно-доминантными мутациями или представлять собою аутосомно-доминантное заболевание в семьях с неполной пенетрантностью.
В некоторых спорадических случаях генетическое заболевание может вообще отсутствовать. Аналогичные дистрофии сетчатки могут развиваться на фоне мутаций митохондриальной ДНК, но при этом наблюдаются и другие системные аномалии.
Для Х-сцепленной и аутосомно-доминантной формы пигментного ретинита характерны более ранний дебют и более тяжелое течение по сравнению с доминантной формой. При диагностике необходимо принимать во внимание клиническую картину, особенно при консультации пациентов с очевидно спорадическим заболеванием. При тяжелом течении болезни у женщины более вероятна аутосомно-рецессивная форма, тогда как при тяжелом поражении у пациента мужского пола вероятнее наличие Х-сцепленной или аутосомно-рецессивной формы заболевания. Однако у значительного числа пациентов пигментный ретинит развивается спорадически и протекает в легкой форме; у некоторых таких пациентов, вероятно, имеются вновь возникшие аутосомно-доминантные мутации.
Перед консультированием важно обследовать других членов семьи, особенно матерей тяжело больных пациентов мужского пола, у которых (матерей) могут обнаружиться изменения глазного дна и электрофизиологические аномалии, указывающее на статус Х-сцепленной гетерозиготы.
Качество генетического консультирования зависит от идентификации патогенной мутации. В этой области достигнут значительный прогресс, в настоящее время возможно проведение генетической диагностики многих форм пигментного ретинита. Идентифицировано более пятидесяти генов, ответственных за развитие несиндромального пигментного ретинита; они кодируют белки каскада фототрансдукции, белки, участвующие в метаболизме витамина А и процессах межклеточного взаимодействия, структурные белки фоторецепторов и факторы транскрипции, внутриклеточные транспортные протеины и факторы сплайсинга.
и) Молекулярная генетика. Аутосомно-доминантный пигментный ретинит. Наблюдается выраженная генетическая гетерогенность аутосомно-доминантного пигментного ретинита. Идентифицированы мутации более двадцати генов, вызывающих приблизительно 60-70% случаев аутосомно-доминантного пигментного ретинита, наиболее частыми из них являются мутации гена родопсина (RHO). Варианты последовательности RP1 и PRPF31 — следующие по частоте причины аутосомно-доминантного пигментного ретинита.
Мутации гена родопсина выявляются приблизительно у 25% пациентов с аутосомно-доминантным пигментным ретинитом, идентифицировано более восьмидесяти различных мутаций. Наблюдается выраженная вариабельность фенотипа глаз при различных мутациях; эти же мутации также выявляются при доминантных формах врожденной стационарной ночной слепоты и аутосомно-рецессивном пигментном ретините. Фенотипическая вариабельность более выражена при мутациях гена PRPH2 (ранее называвшегося RDS/периферии) — клиническая картина может напоминать пигментный ретинит, колбочко-палочковую дистрофию, колбочковую дистрофию, синдром пятнистой сетчатки или макулярную дистрофию. Мутации PRPH2 вызывают примерно 5% случаев аутосомно-доминантного пигментного ретинита.
Известна нетипичная форма пигментного ретинита, наследуемая по дигенному механизму, вызываемая мутациями генов PRPH2 и ROM1 (протеина наружного сегмента палочек 1) в одной семье. Индивиды—носители мутации одного гена, у которых отсутствует мутация второго гена, клинически здоровы. Больные индивиды являются дигетерозиготами, имеющими мутации и ROM1 и PRPH2. Периферии локализуется в дисках наружных сегментов палочек и колбочек, тогда как ROM 1 обнаружен только в палочках. Взаимодействие двух белков в наружных сегментах палочек имеет важное значение для формирования структуры наружного сегмента: вероятно, некоторых мутаций гена PRPH2 недостаточно для развития серьезного поражения фоторецепторов, если эта аномалия не сопровождается дефектом протеина ROM1.
Идентифицируется все больше сплайс-факторов, связанных с аутосомно-доминантным пигментным ретинитом, включая мутации PRPF31, PRPF3, PRPF6 и PRPF8. Дефект сплайсинга может оказаться патогенным только в условиях высокой потребности в сплайсинге. Такие условия существуют в фоторецепторах палочек, где требуется ежедневное восстановление белков дисков, что может объяснять парадокс изолированного поражения сетчатки при повсеместной экспрессии генов сплайс-факторов.
Корреляции генотип-фенотип при аутосомно-доминантном пигментном ретините описаны в литературе раздела медицинская библиотека данного сайта.
к) Аутосомно-рецессивный пигментный ретинит. В настоящее время идентифицированы мутации более тридцати генов, вызывающих приблизительно 40-50% случаев аутосомно-рецессивного пигментного ретинита: наиболее часто это мутации гена USH2A (10-15% случаев аутосомно-рецессивного пигментного ретинита), более тяжелые мутации этого гена вызывают синдром Usher 2 типа.
Идентифицированы мутации генов, кодирующих многие компоненты каскада фототрансдукции, в том числе:
1. Ген родопсина.
2. Гены, кодирующие α- и β-субъединицы палочковой цГМФ-фосфодиэстеразы.
3. Гены α- и β-субъединиц цГМФ-зависимых катионных каналов палочек.
4. Ген SAG, кодирующий аррестин.
5. Гены, кодирующие компоненты зрительного цикла, в том числе участвующие в метаболизме витамина А, также могут участвовать в патогенезе аутосомно-рецессивного пигментного ретинита, в том числе гены RPE65, ABCA4, LRAT и RLBP1.
л) Х-сцепленный пигментный ретинит. При Х-сцепленном пигментном ретините были идентифицированы мутации двух генов, RPGR (RP3) и RP2 (RP2). Варианты последовательности RPGR вызывают приблизительно 75% всех случаев Х-сцепленного пигментного ретинита. Сообщается о трех других локусах: Хр22 (RP23), Xp21.3-p21.2 (RP6) и Xq26-27 (RP24).
Мутации RPGR обычно связаны с типичной палочко-колбочковой дегенерацией, но у нескольких пациентов наблюдались дистрофия сетчатки, глухота и аномалии ресничек респираторного эпителия. Большинство мутаций могут вызывать преждевременную терминацию трансляции. Экзон ORF15 является «горячей точкой» мутаций и его мутации вызывают 80% случаев Х-сцепленного пигментного ретинита. RPGR может выполнять роль регулятора специфического типа мембранного транспорта, особенно активного в сетчатке или ПЭС.
Мутации RP2 вызывают до 15% Х-сцепленного пигментного ретинита. RP2 — недавно открытый протеин, функция которого неизвестна, он метится двойной ацил-модификацией N-конца и транспортируется к плазматической мембране. Дважды ацилированные белки метятся и транспортируются в липидные рафты, что, возможно, указывает на участие этого протеина в сигнальной трансдукции.
м) Ведение пигментного ретинита. Специфической терапии большинства форм пигментного ретинита не существует. При некоторых редких заболеваниях, биохимические механизмы которых изучены лучше, дегенерацию можно замедлить при помощи диеты.
В одном рандомизированном контролируемом исследовании действия витамина А при пигментном ретините выявлено небольшое замедление деградации колбочковой ритмической ЭРГ; однако этот эффект был пограничным и никакого влияния на динамику остроты или поля зрения выявлено не было. Этот вид терапии не получил широкого распространения.
Несмотря на отсутствие эффективного лечения пигментного ретинита, офтальмолог играет важную роль в ведении пациента и оказании помощи его семье. После постановки диагноза важно, чтобы родители и ребенок (если он достаточно взрослый) получили полное и доброжелательное объяснение; их можно заверить, что большинство больных детей заканчивают обычную школу, поскольку центральное зрение страдает позже. Родителей обычно волнует, что у других детей тоже имеется риск развития заболевания; им нужно предложить генетическую консультацию и, вероятно, следует обследовать и других членов семьи. Также полезно иметь адрес ближайших групп взаимопомощи пациентов, например Национальной ассоциации пигментного ретинита.
При проблемах, связанных со зрением, также должна оказываться практическая помощь. Многие пациенты с пигментным ретинитом плохо видят на ярком свету и трудно адаптируются при изменении освещения с яркого на тусклое; им могут оказаться полезными затемненные линзы. Необходима коррекция выраженных аномалий рефракции. При макулярном отеке возможно перорального назначение ацетазоламида, местное применение дорзоламида, введение стероидов внутривенно или в нижнюю стенку глазницы или интравитреально бевацизумаба или ранибизумаба. При развитии отека или макулярной атрофии может потребоваться помощь по поводу слабовидения. Также зрение может ухудшаться вторично вследствие развития задней субкапсулярной катаракты и, хотя операция по поводу катаракты часто успешно выполняется у взрослых с пигментным ретинитом, такая необходимость у детей возникает редко.
н) Прогноз. Прогноз при пигментном ретините зависит от типа заболевания и патогенной мутации. В идеале прогноз должен основываться на идентификации специфической генной мутации. Рекомендации по прогнозу также должны основываться на фенотипе и выставленном диагнозе, данных семейного анамнеза и установленного механизма наследования. Следует обследовать всех больных членов семьи, чтобы оценить динамику тяжести заболевания с возрастом. Однако встречается внутрисемейная вариабельность экспрессии заболевания: исход для зрения у более старших больных родственников не обязательно является надежным прогностическим признаком при консультировании маленьких детей.
При Х-сцепленном пигментном ретините у больных мужчин в раннем детстве развивается ночная слепота, обычно возникают обширные дефекты поля зрения во втором, и утрачивается центральное зрение в третьем десятилетии жизни. К четвертому десятилетию жизни у большинства из них острота зрения не превышает счет пальцев. Аутосомно-рецессивный пигментный ретинит — очень гетерогенное состояние, поэтому точное прогнозирование затруднено. Заболевание обычно рано дебютирует и протекает тяжело. У большинства пациентов во втором десятилетии жизни отмечается тяжелое сужение полей зрения, а к тридцати годам может отмечаться выраженное ухудшение центрального зрения. У некоторых пациентов с рецессивной формой пигментного ретинита заболевание протекает более доброкачественно.
При аутосомно-доминантном пигментном ретините прогноз более благоприятный. Хотя уже в детстве могут развиваться ночная слепота и дефекты поля зрения, центральное зрение может оставаться нормальным на протяжении всей жизни. У многих пациентов до пятого или шестого десятилетия жизни сохраняется достаточно высокая острота зрения, хотя поля зрения могут быть крайне суженными. Однако даже в случаях пигментного ретинита, вызванного мутациями в одном локусе, отмечается выраженная вариабельность. Тяжелые рано дебютирующие формы не характерны.
Истинный секторальный пигментный ретинит характеризуется лучшим среди всех форм заболевания прогнозом. Хотя могут развиваться тяжелые дефекты верхних квадрантов поля зрения, тяжелое поражение макулы развивается нечасто.
«Ковровый (tapetal)» рефлекс глазного дна у женщины — носителя Х-сцепленного пигментного ретинита.
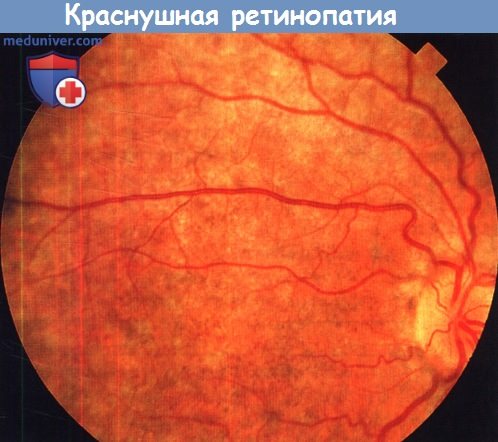
Краснушная ретинопатия, видна пятнистая пигментация сетчатки.
Обычно функции сетчатки не нарушены, на ЭРГ изменения отсутствуют,
тогда как в большинстве случаев дистрофий сетчатки, сопровождающихся глухотой,
при электроретинографии регистрируются тяжелые аномалии.
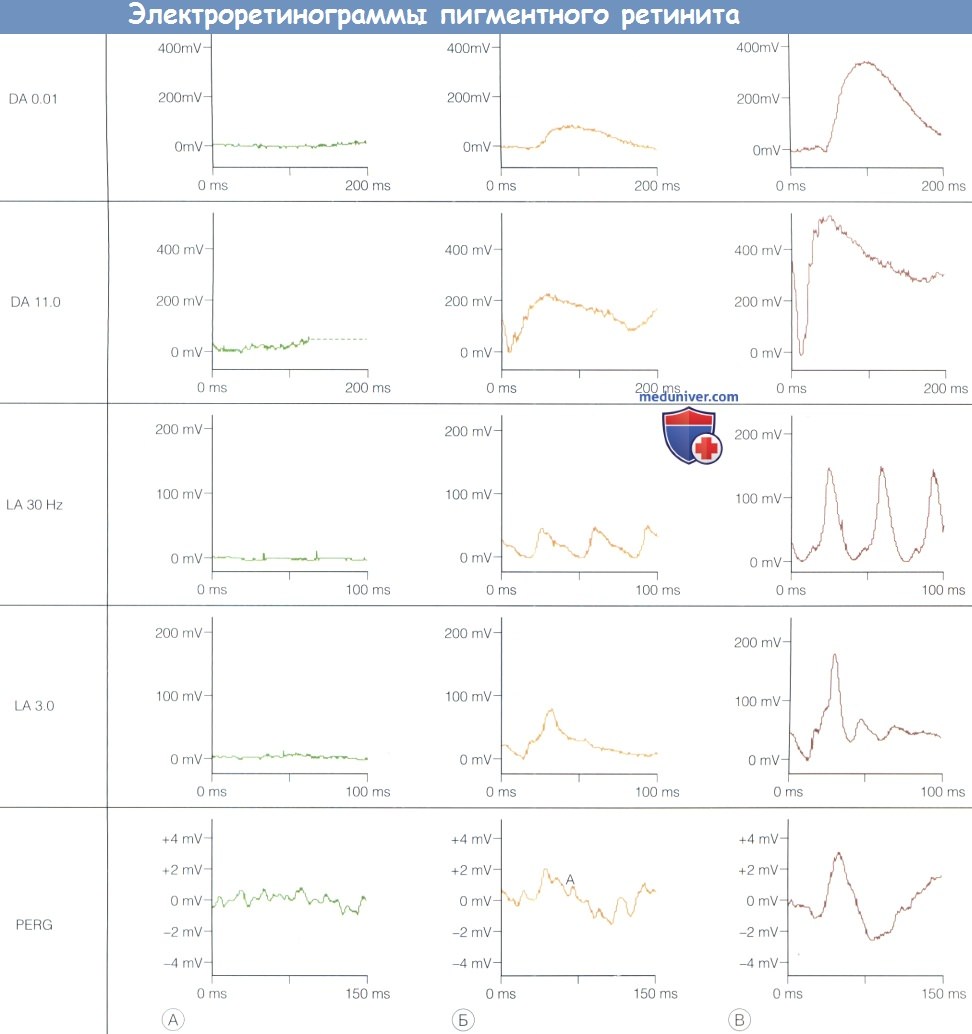
Электроретинограммы маленького пациента с Х-сцепленным пигментным ретинитом (А), его матери-гетерозиготы (Б) и типичная ЭРГ здорового человека (В).
У больного регистрируется лишь остаточная ЭРГ активность. Электроретинограммы его матери несколько субнормальны при любой стимуляции в любом состоянии,
субнормальная а-волна в ответ на стимуляцию яркой вспышкой (DA 11,0) подтверждает наличие аномалий на уровне фоторецепторов.
У гетерозигот обычно выявляются аномалии ЭРГ, которые по тяжести варьируют от легких до тяжелых.