Основные признаки синдрома Барде-Бидля — рано дебютирующие дегенерация сетчатки, ожирение, полидактилия, почечная недостаточность, гипогонадизм и когнитивные нарушения. Вторичные проявления могут включать в себя аносмию, диабет, патологию сердца, фиброз печени, брахидактилию и болезнь Гиршпрунга.
Дистрофия сетчатки дебютирует рано и приводит к инвалидизации по зрению еще до наступления взрослого возраста. Выраженные аномалии ЭРГ могут развиваться уже в трехлетием возрасте. Формальная слепота обычно наступает до начала второго десятилетия жизни.
Дистрофии сетчатки при синдроме Барде-Бидля представляют собою, в основном, палочко-колбочковые или колбочко-палочковые дистрофии, обычно они классифицируются как тотальная дегенерация сетчатки, поскольку поражаются и палочки, и колбочки.
Ожирение — второй основной признак, развивающийся у 72-96% пациентов с синдромом Барде-Бидля, оно обычно начинается в раннем детстве и прогрессирует с возрастом. Ожирение имеет и центральное (контроль аппетита со стороны гипоталамуса) и периферическое (жировая ткань) происхождение. Аномалии конечностей выявляются почти у 95% пациентов с синдромом Барде-Бидля, обычно это постаксиальная полидактилия (69%).
Другие аномалии конечностей, такие как брахидактилия или синдактилия часто выявляются на ладонях или стопах и имеют диагностическое значение. Часто наблюдаются аномалии гениталий: гипогонадизм у мальчиков и атрезия влагалища у девочек. Изредка развивается гидрометрокольпос, мальформация влагалища новорожденных, приводящая к развитию массивного абдоминального образования. В старшем детском возрасте может развиваться дисфункция почек, приводящая к почечной недостаточности.
Нейропсихиатрические нарушения могут включать в себя задержку развития, умственную отсталость, нарушения обучаемости, дефекты речи и поведенческие нарушения. Интеллектуальные функции варьируют от тяжелой умственной отсталости (29%) до низкого или среднего интеллекта (29%). Часто отмечаются замедленное мышление и гиперэмоциональный статус.
Синдром Барде-Бидля — аутосомно-рецессивное гетерогенное заболевание; идентифицировано 16 генов, вызывающих примерно 85% всех случаев. Все гены принимают участие в биогенезе и/или работе ворсинок. Чаще всего выявляются BBS1 и BBS 10, каждый из них вызывает примерно 20% всех случаев заболевания. Встречаемость других генов BBS варьирует от единственной семьи до нескольких процентов семей, наследующих мутации.
По результатам проведенных молекулярных и функциональных исследований представления о классическом механизме аутосомно-рецессивного наследования были пересмотрены, была создана модель олигогенного наследования и влияния на фенотип дополнительных генетических модуляторов.
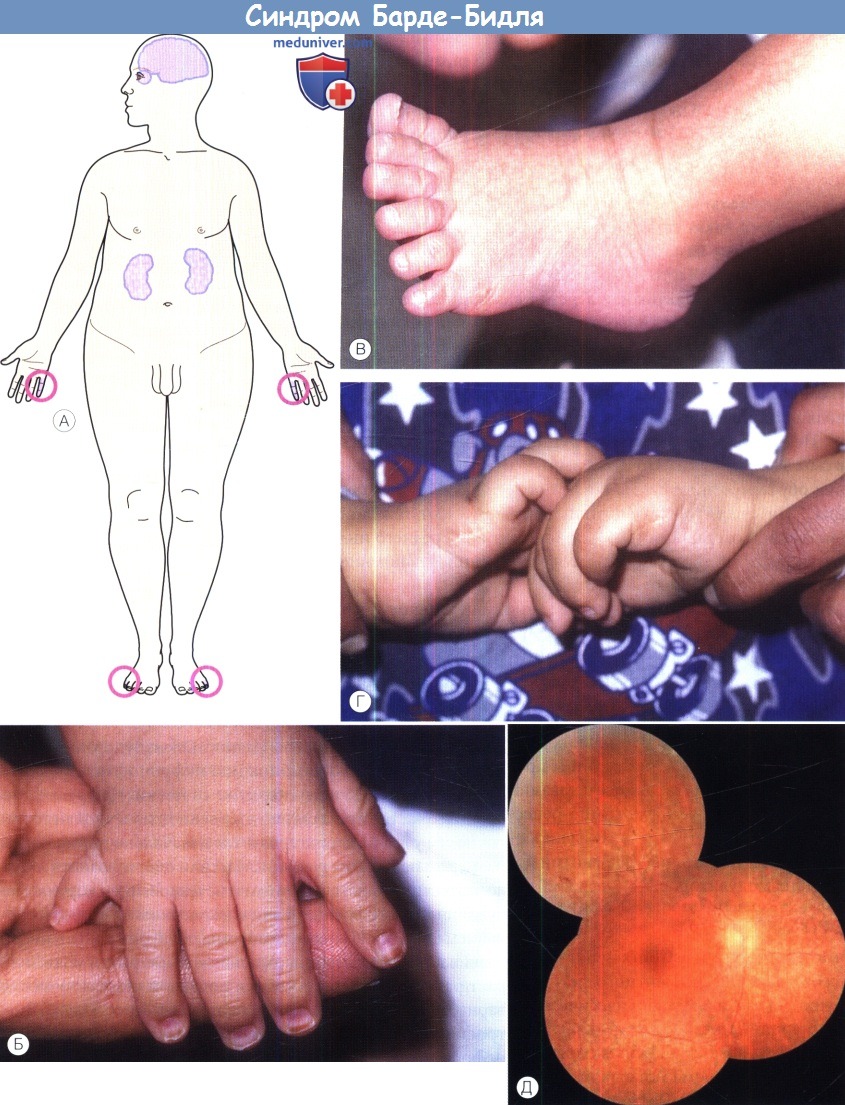
Синдром Барде-Бидля:
(А) Схематическое изображение пяти основных проявлений синдрома Барде-Бидля: ожирение, полидактилия, дисфункция почек, пигментный ретинит и когнитивные нарушения.
(Б) Полидактилия кисти у шестимесячного пациента — носителя мутации гена BBS1.
(В) Полидактилия стопы у шестимесячного пациента — носителя мутации гена BBS1.
(Г) Двусторонние рубцы после хирургического удаления дополнительных пальцев у старшего брата предыдущего пациента.
(Д) На фотографии глазного дна подростка — носителя мутации гена BBS16 видна обширная дегенерация сетчатки.