2. Определения:
• Клинически и генетически неоднородная группа наследственных состояний, характеризующихся фокальным или диффузным утолщением периферических нервов
• В зависимости от преимущественных двигательных и/или чувствительных нарушений подразделяется на три основных категории
о Поражение двигательных и чувствительных нервов → НМСН:
- НМСН I (синдром Шарко-Мари-Тута I типа, ШМТ 1)
- НМСН II (малоберцовая мышечная атрофия, ШМТ 2)
- НМСН III (болезнь Дежерин-Сотта [БДС], гипертрофическая нейропатия новорожденных, врожденная гипомиелинизирующая нейропатия)
о Преимущественно моторные нарушения → дистальная НМН, спинальный вариант ШМТ, дистальная спинальная мышечная атрофия
о Преимущественно периферические чувствительные нарушения ± вегетативные нарушения → наследственная сенсорная нейропатия (НСН) или наследственная сенсорная и автономная нейропатия (НСАН)
б) Визуализация:
1. Общие характеристики:
• Наиболее значимый диагностический признак:
о Фокальное или диффузное утолщение периферического нерва ± атрофия мышц дистальных сегментов конечности
• Локализация:
о Периферические нервы ± интрадуральные сегменты корешков
2. КТ при гипертрофической нейропатии:
• КТ с КУ:
о Утолщение ± контрастирование нервов
3. МРТ при гипертрофической нейропатии:
• Т1-ВИ:
о Веретеновидные гипоинтенсивные образования периферических нервов ± корешков конского хвоста
о ± патологическая гиперинтенсивность сигнала мышц, уменьшение их объема (хроническая денервация → жировая атрофия)
• Т2-ВИ:
о Веретеновидные гипоинтенсивные образования периферических нервов ± корешков конского хвоста
о Нарушение нормальной фасцикулярной архитектуры измененного сегмента нерва
о ± патологическая гиперинтенсивность мышц, признаки отека на FS Т2-ВИ (острая денервация)
• STIR:
о Изменения аналогичны FS Т2-ВИ
• Т1-ВИ с КУ:
о ± патологическое контрастное усиление пораженных нервов (чаще всего при НМСН I и III)
4. Несосудистые интервенционные рентгенологические исследования:
• Миелография:
о ± утолщение корешков, миелографический блок вследствие стеноза позвоночника
5. Рекомендации по визуализации:
• Наиболее оптимальный метод диагностики:
о МРТ высокого разрешения (МР-нейрография)
• Протокол исследования:
о Аксиальные Т1-ВИ, FST2 или STIR, FS T1 с КУ
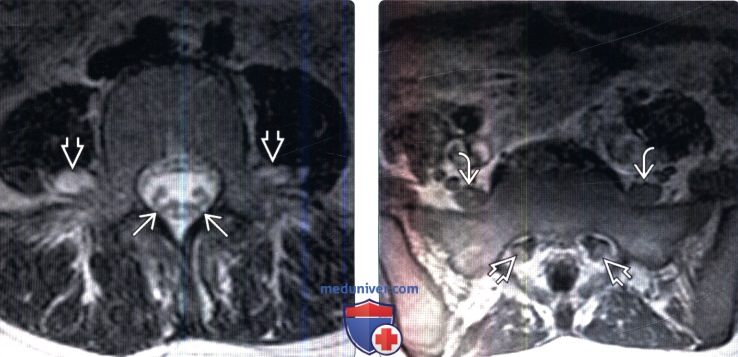
(Слева) На аксиальном Т2-ВИ (Болезнь Шарко-Мари-Тута 1) отмечается легкое диффузное утолщение интрадуральных сегментов корешков конского хвоста, а также двустороннее утолщение экстрадуральных сегментов нервов ЕВ поясничного сплетения медиальней поясничных мышц.
(Справа) Аксиальный срез, Т1-ВИ с КУ (ШМТ 1): на уровне крестца определяется патологические усиление сигнала и утолщение экстрадуральных сегментов крестцовых корешков с обеих сторон и стволов пояснично-крестцового сплетения.
1. Синдром Гийена-Барре:
• Остро развивающийся восходящий паралич с относительным сохранением чувствительности
• Характерное развитие и прогрессирование симптоматики
• Контрастное усиление мягких мозговых оболочек и корешков, как и при ХВДП
2. Хроническая воспалительная демиелинизирующая полинейропатия (ХВДП):
• Повторные эпизоды демиелинизации, ремиелинизации → утолщение периферических и спинномозговых нервов с формирование картины «луковой кожуры»
• МР-картина напоминает НМСН, плексиформную нейрофиброму
• От НМСН помогает отличить наличие воспалительного инфильтрата (гистология)
3. Опухоль оболочки нерва:
• Шваннома, солитарная нейрофиброма, плексиформная нейрофиброма:
о Диффузное утолщение корешка, контрастное усиление о ± кожные стигмы при НФ1
о Генетическое консультирование, характерные клинические признаки, которые позволяют отличить опухоль от НМСН и ХВДП
4. Тракционное повреждение нерва:
• Утолщение нерва, искажение его внутренней архитектуры
• Травма в анамнезе и характерные (травматические) изменения тканей
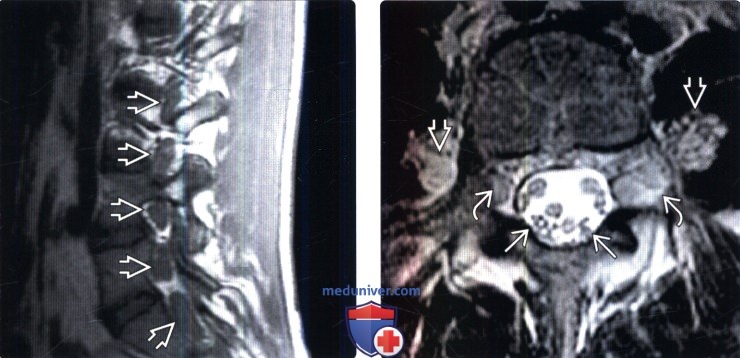
(Слева) На сагиттальном Т1-ВИ (ШМТ) на уровне невральных отверстий поясничного отдела позвоночника отмечается патологическое утолщение экстрадуральных сегментов спинномозговых нервов в невральных отверстиях.
(Справа) На аксиальном Т2-ВИ (ШМТ) отмечается патологическое утолщение корешков конского хвоста, а также утолщение и патологическая гиперинтенсивность сигнала фораминальных и паравертебральных сегментов спинномозговых нервов.
г) Патология:
1. Общие характеристики:
• Этиология:
о НМСН I (ШМТ 1): нарушение миелинизации периферических нервов:
- Мутация гена периферического миелинового протеина-22 (РМР-22) → образование нестабильного миелина, который подвергается спонтанной демиелинизации
- Пролиферация шванновских клеток и ремиелинизация аксонов
- Повторные циклы демиелинизации и ремиелинизации приводят к образованию вокруг аксонов концентрических слоев миелина (картина «луковой кожуры»)
о НМСН II (ШМТ 2): первичное поражение аксонов, валлерова дегенерация без циклов демиелинизации/ремиелинизации
о НМСН III (болезнь Дежерин-Сотта): тяжелая сегментарная де-миелинизация, истончение миелиновых оболочек
• Генетика:
о НМСН IA (ШМТ 1 А): аутосомно-доминантный тип наследования, короткое плечо 17 хромосомы
о НМСН IB (ШМТ 1 В): длинное плечо 1 хромосомы
о НМСН II: 1 хромосома
о НМСН III: аутосомно-рецессивный тип наследования (спорное утверждение), 17 хромосома
2. Стадирование, степени и классификация гипертрофической нейропатии:
• НМСН типы I A, IB (ШМТ 1 А, 1 В)
• НМСН тип II (малоберцовая мышечная атрофия нейронального типа)
• НМСН тип III (гипертрофическая нейропатия новорожденных, болезнь Дежерин - Сотта)
• НМСН тип IV (гипертрофическая нейропатия, связанная с избытком фитановой кислоты)
• НМСН тип V (сочетание со спастической параплегией)
• НМСН тип VI (сочетание с атрофией зрительных нервов)
• НМСН тип VII (сочетание с пигментным ретинитом)
3. Макроскопические и хирургические особенности:
• Гипертрофированные утолщенные корешки спинного мозга, периферические нервы
• Во время операции может оказаться, что корешки полностью заполняют собой дуральный мешок
4. Микроскопия:
• Гистологические признаки демиелинизации, ремиелинизации:
о НМСН I: первичная патология миелиновых оболочек → демиелинизация/ремиелинизация, формирование концентрических «луковиц», положительных на протеин S100, вторичные изменения аксонов
о НМСН II: первичная патология аксонов, их повреждение и валлерова дегенерация
о НМСН III: тяжелая гипомиелинизация/демиелинизация, повреждение аксонов, истончение миелиновых оболочек
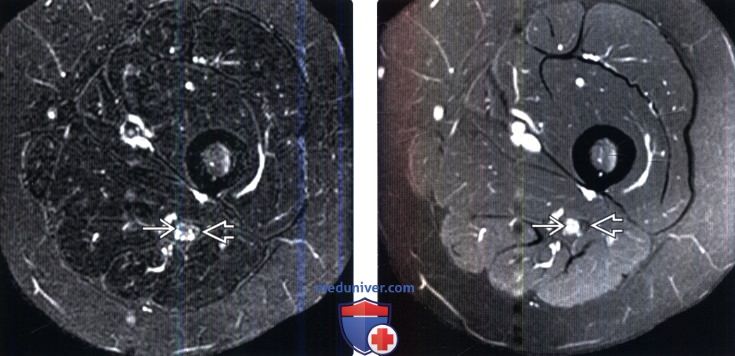
(Слева) Сагиттальный срез, STIR МР-И (неизвестный тип НМСН, радикулопатия S1): гипоинтенсивное центральное новообразование большеберцовой порции седалищного нерва, оттесняющее гиперинтенсивные пучки нервных волокон к периферии. Малоберцовая порция нерва выглядит нормально.
(Справа) На аксиальном FS Т1-ВИ с КУ (неизвестный тип гипертрофической нейропатии, радикулопатия S1) отмечается очень умеренное патологическое контрастное усиление большеберцовой порции седалищного нерва. Контрастного усиления соседней малоберцовой порции не отмечается.
е) Диагностическая памятка:
1. Клиническая картина:
• Наиболее распространенные симптомы/признаки:
о НМСН I:
- Слабость/атрофия мышц дистальных сегментов конечностей (моторные > сенсорные нарушения), деформации стоп (pes cavus, молоткообразные пальцы), снижение сухожильных рефлексов, ± пальпируемые объемные образования нервов
- Относительно отсутствие болевого синдрома (в отличие от приобретенных нейропатий)
- Часты аналогичные заболевания у родственников
о НМСН II: часто минимально выраженная симптоматика
о НМСН III: симптоматика похожа, но выражена в большей степени, чем при НМСН I, более ранее развитие симптоматики:
- Отсроченные моторные нарушения, прогрессирующая слабость мышц нижних и верхних конечностей
• Другие симптомы/признаки:
о ± пальпируемые объемные образования нервов
о Боль в спине/корешковый болевой синдром ± миелопатия
о Чувствительные нарушения, локальная болезненность, дизестезии
о Холодные стопы, выпадение волос, отек нижних конечностей
• Особенности клинического течения
о НМСН тип I: слабость, атрофия мышц дистальных сегментов конечностей (нижние > верхние)
- Гипорефлексия или арефлексия, ортопедические деформации стоп, сколиоз (37-50%), тремор (< 25%)
о НМСН тип II: периферические нервы клинически не утолщены, слабость нижних конечностей > кистей, чувствительные нарушения в дистальных сегментах конечностей, деформации стоп (менее выраженные, чем при НМСН I)
о НМСН тип III: новорожденные с гипорефлексией, пальпируемое утолщение периферических нервов, чувствительные нарушения ± атаксия
2. Демография:
• Возраст:
о НМСН I: манифестация в первом десятилетии жизни, у некоторых пациентов манифестация только в молодом или среднем возрасте
о НМСН II: нередко диагностируется уже в зрелом возрасте, первые симптомы обычно появляются в течение второго десятилетия жизни
о НМСН III: манифестация обычно в грудном или раннем детском возрасте
• Пол:
о М=Ж
• Эпидемиология:
о НМСН I: частота 1 5 случаев на 1 00000
- ШМТ 1 А: 10,5 на 100000 (70% случаев НМСН I типа)
о НМСН II: частота семь случаев на 100000
о НМСН III: редкая патология
3. Течение заболевания и прогноз:
• Нормальная продолжительность жизни
• Медленно прогрессирующее заболевание, выраженность функциональных нарушений варьирует в достаточно значительных пределах
4. Лечение гипертрофической нейропатии:
• Консервативное: профилактика травм (изменение образа жизни, профилактика травм, связанных с родом занятий), лечение приобретенных нейропатий, физиотерапия, ортезирование стопы и голеностопного сустава, противовоспалительные препараты
• Хирургическое: коррекция ортопедических деформаций
ж) Диагностическая памятка:
1. Следует учесть:
• Пациентам с веретеновидным поданным МРТ утолщением нервов рекомендуется генетическое консультирование
2. Советы по интерпретации изображений:
• При обнаружении при МР-исследовании патологически утолщенных периферических нервов следует думать о НМСН
з) Список использованной литературы:
1. Pitarokoili К et al: Nerve ultrasound in a case of multifocal motor neuropathy without conduction block. Muscle Nerve. ePub, 2015
2. Goedee SH et al: Distinctive patterns of sonographic nerve enlargement in Charcot-Marie-Tooth type 1A and hereditary neuropathy with pressurepalsies. Clin Neurophysiol. ePub, 2014
3. Berciano J et al: CMT1A duplication: refining the minimal adult phenotype. J Peripher Nerv Syst. 1 3(4):31 0-2, 2008
4. Chung KW et al: Different clinical and magnetic resonance imaging features between Charcot-Marie-Tooth disease type 1A and 2A. Neuromuscul Disord. 18(8):610-8, 2008
5. Gallardo E et al: Magnetic resonance imaging findings of leg musculature in Charcot-Marie-Tooth disease type 2 due to dynamin 2 mutation. J Neurol. 255(7/986-92, 2008
6. Somasundaram S et al: Hereditary spastic paraplegia with a thin corpus callosum. Pediatr Radiol. 37(5):503-5, 2007
7. Berciano J et al: Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 77(10):1169-76, 2006
8. Chung KW et al: Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. Brain. 1 29(Pt 8):2103-18, 2006
9. Ellegala DB et al: Characterization of genetically defined types of Charcot-Marie-Tooth neuropathies by using magnetic resonance neurography. J Neurosurg. 1 02(2/242-5, 2005
10. Kassubek J et al: Corticospinal tract MRI hyperintensity in X-linked Charcot-Marie-Tooth Disease. J Clin Neurosci. 12(5):588-9, 2005
11. Kretzer RM et al: Hypertrophic neuropathy of the cauda eguina: case report. Neurosurgery. 54(2/51 5-8; discussion 518-9, 2004
12. Liao JP et al: Nerve root hypertrophy in CMT type 1 A. Neurology. 62(5/783, 2004
13. Moore KR et al: The value of MR neurography for evaluating extraspinal neuropathic leg pain: a pictorial essay. AJNR Am J Neuroradiol. 22(4):786-94, 2001