Бластная NK-клеточная лимфома встречается редко и, как правило, у пожилых людей. Обычно опухоль выявляется в виде кожных узелков. Часто поражаются лимфатические узлы, селезенка и костный мозг, и к моменту установления диагноза заболевание может быть диссеминированным, с наличием злокачественных клеток в периферической крови и панцитопепией. Есть сведения о том, что это заболевание может быть гетерогенным с кожными и нскожными подтипами (Suzuki и соавт., 2005).
Несмотря на первоначальный положительный ответ на химиотерапию, прогноз неблагоприятный, часто возникают рецидивы с поражением нейтральной нервной системы.
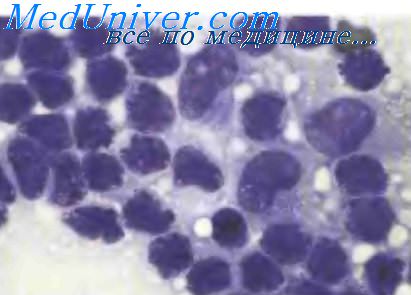
Для поражения кожи и других органов характерна инфильтрация довольно одинаковыми клетками средних размеров. В небольшом количестве случаев состав клеточного инфильтрата полиморфный, с наличием более крупных форм, что напоминает личфобластную лимфому или острый миелоидный лейкоз, вплоть до вариантов single-file или «Indianfile». Хроматин распределен равномерно (рассеянный тип) или по типу «кружев»; ядрышки неразличимы.
Несмотря на то, что этот вариант в классификации ВОЗ описан как бластная NK-клеточная лимфома, клеточное происхождение остается неопределенным. Имеются сведения о происхождении этой опухоли из дендритных плазмоцитоидных моноцитов 2 типа.
Характерна позитивность опухолевых клеток для CD4 и CD56, что объясняет происхождение одного из альтернативных терминов - «агранулярная CD4 +, CD56+ гематодермная опухоль» (Petrclla и соавт., 2002) Обычные лимфоидные и миелоидные маркеры (включая CD3 и CD5) отсутствуют. Характерен следующий иммунофепотип: CD45 — слабо позитивный. CD45RA — позитивный, CD123 — позитивный. CD38, CD68 и CD7 — обычно позитивные.
Диагноз бластной NK-клеточной лимфомы следует ставить при выявлении характерною иммунофенотипа и возможности исключения лимфобластной лимфомы или миелоидного лейкоза. Для подтверждения отсутствия перегруппировки TCR гена можно использовать ГЩР.