Нервная система при врожденных дефектах метаболизма ГАМК
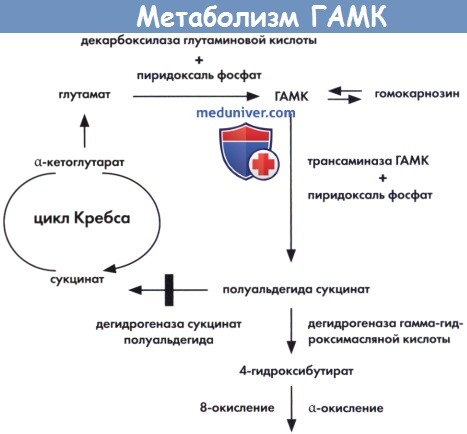
Гамма-аминомасляная кислота (4-аминомасляная кислота), основная аминокислота, являющаяся ингибирующим нейромедиатором, синтезируется из глутаминовой кислоты с помощью глутамат декарбоксилазы. Катаболизм включает трансаминирование, в результате которого образуется сукцинат полуальдегида, и окисление до сукцината, который в итоге участвует в цикле Кребса. Пиродоксальфосфат является коферментом для всех трех реакций. В рамках второго пути, катализируемого дегидрогеназой, происходит превращение сукцината полуальдегида в гамма-гидрокисибутират, который в дальнейшем окисляется.
Кроме того, основным предшественником глутамата в нейронах является глутамин, который синтезируется в глиальных клетках с помощью глутаминсинтетазы. Глутамин затем транспортируется в нейроны, где метаболизируется глутамат, а затем ГАМК. Данный челночный механизм между нейронами и глиальными клетками позволяет поддерживать существование данной системы. Гомокарнозин является дипептидной формой ГАМК. Чаще всего дефект метаболизма ГАМК отмечается на уровне сукцинил полуальдегид дегидрогеназы. Дефицит ГАМК трансаминазы и гомокарнозиноз являются очень редкими нарушениями. Дефект синтеза глутамина является новым заболеванием, описанным у новорожденных с тяжелыми мальформациями головного мозга.
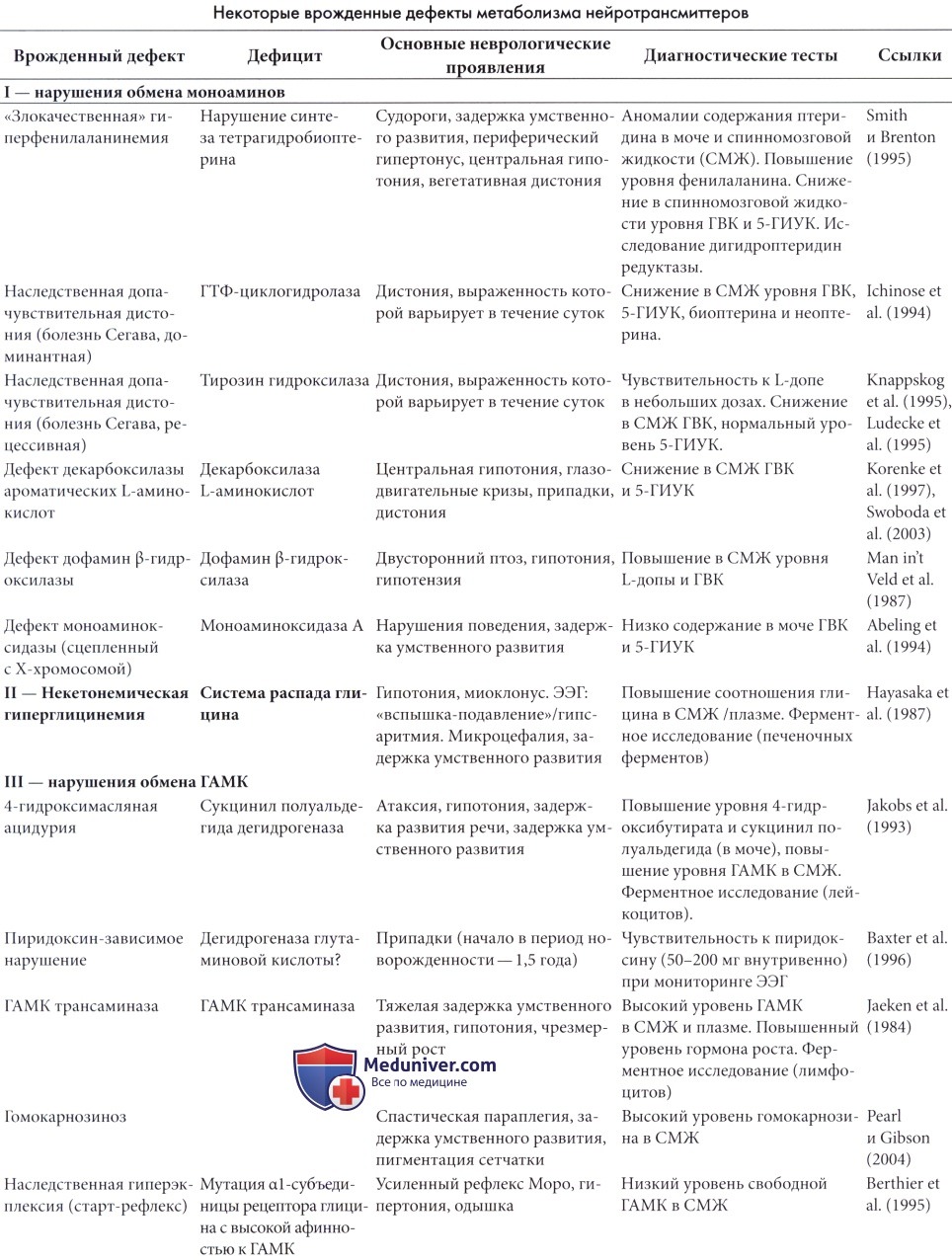
а) 4-гидроксимасляная ацидурия. 4-гидроксимасляная ацидурия является наиболее частым нарушением обмена ГАМК. Заболевание наследуется аутосомно-рецессивным путем и характеризуется накоплением гамма-гидроксимасляной кислоты (ГГМ) и ГАМК в жидкостях организма вследствие дефицита сукцинил полуальдегид дегадрогеназы (СПАДГ). Дефицит СПАДГ выявлен в лимфоцитах и фибробластах. У пациентов идентифицирован ген СПАДГ и множество его мутаций. Пренатальная диагностика может основываться на оценке накопления ГГМ в амниотической жидкости, определении ферментной активности в культуре амниоцитов или молекулярном анализе ДНК хорионических ворсин (Gordon, 2004).
1. Патофизиология. Данное заболевание приводит к накоплению ГГМ и ГАМК в сочетании с низким уровнем глутамата в спинномозговой жидкости. ГГМ является нейрофармакологически активным веществом, которое может играть роль нейротрансмиттера или нейромодулятора в головном мозге. В физиологической концентрации ГГМ взаимодействует с ГГМ-рецепторами. В высокой концентрации ГГМ может являться причиной нарушения нормального баланса между глутаминэргическим возбуждающим действием и ГАМК-эргическим ингибированием. Данный дисбаланс может быть вызван антагонистическим воздействием ГГМ на ГАМКB-рецептор и разобщением содержания глутамина и ГАМК. Кроме того, метаболиты, образованные путем окисления ГГМ, могут влиять на продукцию энергии и приводить к окислительному повреждению.
И наконец, ГГМ оказывает отрицательное воздействие на обмен катехоламинов с повышением ГВК и 5-ГИУК (Pearl et al., 2003; Buzzi et al., 2006).
2. Клинические проявления. Известно около сотни случаев заболевания с вариабельным клиническим фенотипом (Pearl et al., 2003; Gordon 2004). Наиболее частыми клиническими проявлениями является задержка моторного, речевого и интеллектуального развития. Часто описываются мышечная гипотония, арефлексия и непрогрессирующая атаксия. Другие проявления включают припадки, поведенческие проблемы, диспраксию глаз, макроцефалию, миопатию, хореоатетозы, нистагм и телеангиэктазии конъюнктивы. На ЭЭГ отмечаются неспецифические признаки с различными фоновыми аномалиями и эпилептиформными проявлениями. При нейровизуализации регулярно обнаруживаются аномалии сигнала бледного шара, подкоркового белого вещества и изредка ствола мозга и зубчатого ядра.
Магнитно-резонансная спектроскопия позволяет выявить повышение ГАМК и метаболитов ГАМК (Novotny et al., 2003).
Метаболизм гамма-аминомасляной кислоты (ГАМК).
3. Биохимическая диагностика. Диагноз подтверждается при выявлении высокого уровня ГГМ в плазме, спинномозговой жидкости и моче в сочетании с нарушением экскреции с мочой некоторых побочных продуктов ГГМ. В спинномозговой жидкости отмечается значимое повышение уровня ГГМ, свободной ГАМК и гомокарнозина. Кроме того, отмечается повышение уровня конечных продуктов обмена дофамина (Pearl et al., 2003).
4. Лечение. Наиболее широко распространенным препаратом для лечения данного заболевания является вигабатрин, необратимый ингибитор ГАМК-трансаминазы. Тем не менее, его долгосрочная эффективность ограничена, и препарат может быть противопоказан в связи с токсическим воздействием на сетчатку. Другие препараты исследуются на мышах с выключенным геном. Бензиодиазепины могут применяться для контроля тревожности и агрессивности. Для контроля припадков противопоказано применение вальпроатов, так как они могут ингибировать остаточную ферментную активность СПАДГ.
б) Зависимость от пиридоксина. Зависимость от пиридоксина является редким заболеванием, характеризующимся неконтролируемыми припадками, которые купируются только при введении пиридоксина. Заболевание наследуется аутосомно-рецессивным путем.
Долгое время считалось, что заболевание является результатом аномалий глутамат декарбоксилазы, но данная гипотеза не подтвердилась. Недавно было продемонстрировано, что зависимость от пиридоксина связана с дефектом специфической дегидрогеназы, антиквитина, необходимой для катаболизма лизина и пепеколина в головном мозге. Нарушение активности антиквитина приводит к накоплению альфа-аминоадипинового полуальдегида (а-ААПА) и пиперидин-6-карбоновой кислоты (П6К). П6К конденсируется с пиридоксальфосфатом, приводя к его инактивации. Ген антиквитина (ALDH7A1) клонирован и выявлены различные его мутации в нескольких семьях (Plecko et al., 2000; Mills et al., 2006).
Столь необычный дефицит пиридоксальфосфата может приводить к снижению уровня ГАМК в головном мозге. Клинические проявления описаны в отдельной статье на сайте.
Диагностика возможна на основании повышения уровня альфа-аминоадипинового полуальдегида в моче (Bok et al., 2007).
в) Дефицит синтеза глутамина. Глутаминсинтетаза (ГС) катализирует превращение глутамата и аммония в глутамин. Высокая активность ГС отмечается в печени, головном мозге и мышцах. Роль глутамина зависит от основной функции органа. В головном мозге синтез глутамина оказывает нейропротективное действие за счет удаления аммония и глутамата. Кроме того, глутамин играет решающую роль в переносе ГАМК, для которого требуется нормальная активность ГС в глиальных клетках. Недавно было описано два случая дефицита ГС у неродственных новорожденных (Haberle et al., 2005,2006). При пренатальной ультразвуковой диагностике были выявлены аномалии, включавшие увеличение желудочков и паравентрикулярные кисты у одного пациента и расширение задней черепной ямки у другого.
При рождении у обоих детей отмечалось очень плохое неврологическое состояние из-за обширной гипотонии и припадков. На МРТ выявлена атрофия с практически полной агирией и паравентрикулярными кистами. У одного пациента отмечались другие дисморфические признаки, он умер в возрасте двух дней. У второго пациента развилась некротическая эритремия, он умер в возрасте четырех недель. При исследовании метаболизма было выявлено заметное снижение уровня глутамина в плазме, моче и спинномозговой жидкости. Последующие исследования показали низкую активность ГС и мутации гена, кодирующего ГС.
г) Другие заболевания, связанные с ГАМК. Дефицит ГАМК трансаминазы и гомокарноциноз были описаны у небольшого количества пациентов (Pearl и Gibson, 2004). Мутации ГАМК рецептора в редких случаях могут приводить к наследуемой доминантным путем идиопатической эпилепсии, связанной с мутацией гена, кодирующего γ2-субъединицу ГАМКA-рецептора (Baulac et al., 2001).
Дефект митохондриального транспортера глутамата может приводить к тяжелой миоклонической эпилепсии новорожденных с паттерном «вспышка-подавление» и пигментным ретинитом. Данное аутосомно-рецессивное заболевание нарушает митохондриальный транспорт глутамата в головном мозге (Molinari et al., 2005).