Термин «спинальный дизрафизм» используется в отношении гетерогенной группы спинальных мальформаций объединенных наличием несовершенного формирования срединной мезенхимальной, костной и невральной структур. Несмотря на снижение частоты проблема по-прежнему не теряет актуальности, предположительно затрагивая 300000 человек в мире. Почти во всех случаях спинальный дизрафизм проявляется расщелиной позвоночника, где дизрафизм костных структур является результатом неполного закрытия спинного канала. Существует мнение, что причиной в некоторых случаях является скорее вторичный разрыв, нежели отсутствие закрытия.
а) Spina bifida cystica. Spina bifida cystica — самый распространенный тип спинального дизрафизма и включает миелошизис, миеломенингоцеле и менингоцеле. За исключением последнего, нервные структуры подвержены внешнему воздействию без кожного покрова. При spina bifida occulta невральные элементы скрыты под кожей и не выпячиваются над уровнем спины.
Миеломенингоцеле и миелошизис, составляющие 95% случаев явного спинального дизрафизма, в основном идентичны, но отличаются тем, что миелошизис — плоский, а миеломенингоцеле — выбухающий. В обоих случаях над спинным мозгом нет мезенхимы, которая остается плоской со срединным желобом, соответствующим открытому эпендимальному каналу. Кожа является прямым продолжением мягких мозговых оболочек, которые присоединяются по боковым краям пластинки (Botto et al., 1999). Возможно сочетание с различными аномалиями. Диастематомиелия имеется в 31-46% случаев, верхнее относительно целе расщепление в 31%, на уровне целе в 22% и каудальнее него в 25% случаев. У некоторых пациентов целе располагается в одной из половин спинного мозга или вовлекает обе половины на разных уровнях, создавая очень асимметричные клинические симптомы.
Гидросирингомиелия присутствует в 33-75% случаев и часто сопровождается тяжелым сколиозом. Гидроцефалия встречается в 90% случаев, нередко с перинатальным началом (Girard et al., 2003, Biggio et al., 2004, Wyldes и Watkinson, 2004).
Гидромиелия, как правило, распространяется по всей длине спинного канала от уровня задвижки (obex) вниз по центральному каналу до верхнего полюса невральной пластинки. ЦСЖ может оставаться в пределах центрального канала или проникать в ткань спинного мозга, поэтому различие между сирингомиелией и гидромиелией несколько надуманно (Breningstall et al., 1992). Во всех случаях спинной мозг прикрепляется в низком положении. Мозговые оболочки чрезвычайно тонкие и легко рвутся, что приводит к высокому риску инфицирования. В нелеченных случаях над менингеальными оболочками и невральной пластинкой происходит эпителизация. Процесс оставляет рубцы с фиброзными зонами перегиба и сдавливанием спинного мозга. В некоторых случаях эпидермальные или дермоидные кисты возникают в результате включения клеток эпидермиса в рубцы, часто сопровождающегося небольшими липомами, лучше видимыми на МРТ, чем на КТ. Возможен сегментарный инфаркт позвоночника. Более, чем в половине случаев миелоцеле бывают поясничными или тораколюмбальными и люмбосакральными в 25%. Цервикальная или торакальная локализация вместе дают около 11% (Matson, 1969).
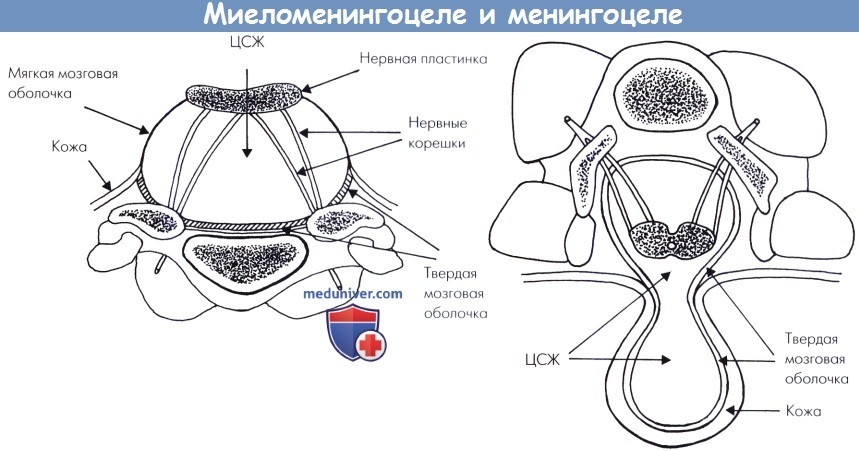
(слева) Миеломенингоцеле. Нервная пластинка не претерпевает нейруляцию и находится в прямой связи с остальной частью дорсальной эктодермы.
Тонкая мембрана, представляющая мягкую мозговую оболочку и легко рвущаяся, связывает пластинку с кожей.
Обратите внимание на отсутствие задней арки позвонка и любых форм мезенхимальных производных позади нервной ткани.
(справа) Менингоцеле. Спинной мозг полностью сформирован. Мозговые оболочки покрыты кожей.
В отдельных случаях, нервные корешки могут иметь патологическое направление и проникают в менингоцеле, выходя из спинального канала, который остается непокрытым мезенхимой позади нервной ткани.
б) Краниальные аномалии часто связаны с миеломенингоцеле, особенно порок развития Киари II, присутствующий в 70% случаев.
Мальформация Киари I, с другой стороны, не связана со spina bifida и редко симптоматичная в детстве (Tubbs et al., 2003а). Патология может быть связана с пороком развития основания черепа и верхнего шейного отдела позвоночника (глава 5) и почти в половине случаев с сирингомиелией. Диагноз возможен и с помощью ультрасонографии (Govaert и de Vries, 1997), но более точно устанавливается при МРТ, с визуализацией низкого положения и удлинения нижних отделов ствола мозга (Mikulis et al., 1992). Симптомы могут появиться в связи со сдавлением мозга и провоцироваться травмой. В различных вариантах они могут включать головную боль, дисфункцию нижних черепных нервов, мозжечковые признаки и боль в шее и затылочной области. Иногда наблюдаются сонливость и эпизоды апноэ (Keefover et al., 1995, Yglesias et al., 1996). Лечение симптоматических случаев заключается в хирургической декомпрессии задней черепной ямки и дает удовлетворительные результаты (Navarro et al., 2004, Schijman и Steinbok, 2004).
Мальформация Киари II представляет собой наиболее частый вариант порока развития Клеланда-Киари (Саша et al., 1995, Stevenson, 2004). Происходит частичная дислокация продолговатого мозга и мозжечка, особенно червя, в цервикальный канал со сжатием верхнего отдела спинного мозга. Дорсальная сторона продолговатого мозга формирует петлю в бульбоспинальном соединении. Мозжечковая ткань может спуститься до уровня СЗ. Четвертый желудочек вытягивается и частично образует грыжевое выпячивание в цервикальном канале. Часто имеется дефект в задней арке первого шейного позвонка с фиброзной спайкой на уровне большого затылочного отверстия, что может сдавливать продолговатый мозг. Задняя черепная ямка маленького размера, с неровностями и эрозиями заднемедиальной части пирамиды височной кости. Так называемый «лакунарный» череп (Luckenschadel) присутствует в 85% случаев с мальформацией Киари II. Это не является прогностическим признаком низкого интеллекта. Ствол мозга имеет удлиненную форму и холмики четверохолмия соединены и имеют форму крючка. Полушария мозжечка могут развиваться кпереди, окружая ствол мозга. Отмечается большой объем промежуточного мозга и нередко гетеротопии полушарного серого вещества.
Часто сообщают о микрогирии, но микроскопическое исследование выявляет нормальное строение коркового вещества, поэтому проявление в большей степени соотносится с механическими факторами, чем с истинными аномалиями. Серповидный отросток мозга и намет мозжечка гипоплазированы. Нарушения нервной системы могут сопровождаться экстрадуральными мальформациями двенадцатиперстной кишки, сердца и пищевода. Более 90% случаев мальформации Киари II связаны с гидроцефалией. В ряде публикаций гидроцефалия была обусловлена стенозом водопровода в 30-73% случаев. В остальных случаях причина была неясна, хотя возможна окклюзия отверстия Мажанди. Еще одним вероятным механизмом может быть дисбаланс продукции ЦСЖ и ресорбции. Возможна гипертрофия сосудистого сплетения. Сама по себе массивная гипертрофия может вызывать изоляцию бокового желудочка с последующей локализованной гидроцефалией (Chadduck и Glasier, 1989). Желудочковая дилятация наблюдается с рождения у 50-75% новорожденных, позволяя предположить, что очевидное развитие гидроцефалии вследствие репарации миеломенингоцеле, возможно, не связано с операцией.
Механизм мальформации Киари II неизвестен. Нисходящая тракция фиксированного спинного мозга уже не считается приемлемым обоснованием. В качестве основного фактора предполагается утечка ЦСЖ во время беременности (Boltshauser, 2004 с гидростатическим нарушением равновесия, приводящим к смещению структур задней ямки в верхние отделы спинномозгового канала. Опускание структур задней ямки встречается только в поздний период беременности, что позволяет предположить возможность предупреждения при внутриутробном хирургическом вмешательстве. Marin-Padilla (1991) полагает, что первичный дефект заключается в развитии мезенхимы цефалических сомитов с гипоплазией основания клиновидной кости, что приводит к формированию маленькой задней черепной ямки.
Различные аномалии, которые сопровождают деформацию Киари II, можно выявить при КТ или (лучше) при МРТ. Полное исследование повреждений спинного мозга (Altman и Altman, 1987, Davis et al., 1988) не требуется перед первичным восстановлением, кроме случаев очевидных асимметричных признаков. Но это обязательно при планировании повторной операции у ранее пролеченного пациента или при вторичных осложнениях. При МРТ точно определяется взаимосвязь кожи и нервной пластинки, расположение нервных стволов и патологические изменения спинного мозга, такие как диастематомиелия и гидромиелия.
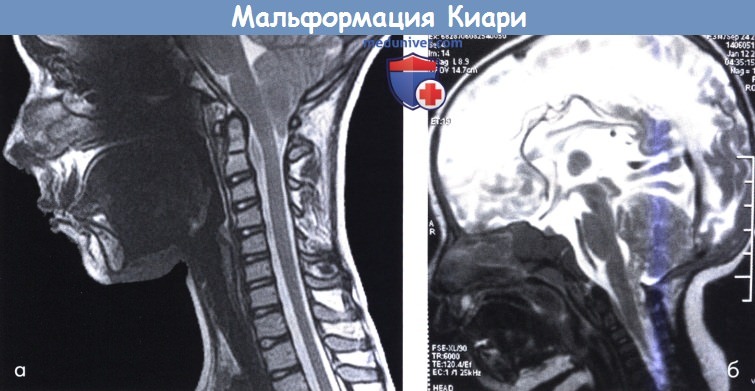
(а) Мальформация Киари I. Дорсальное продолжение миндалин мозжечка ниже уровня большого затылочного отверстия.
Нижняя часть миндалин мозжечка входит в спинномозговой канал ниже уровня затылочной кости.
(б) Мальформация Киари II. Мозжечковая ткань спускается в спинномозговой канал до второго шейного позвонка.
Обратите внимание на удлинение ствола мозга и увеличение межталамической спайки.
Клинические проявления и осложнения spina bifida cystica. При рождении дефект имеет мешковидную структуру, покрытую тонкой мембраной, которая часто разрывается с утечкой ликвора. Окружающая кожа часто с ангиоматозом. Более старые нелеченные повреждения могут частично эпителизироваться. Неврологические проявления миеломенингоцеле включают:
1) прямые последствия спинальной мальформации;
2) последствия гидроцефалии и аномалии ромбовидного мозга; и
3) таковые при сопутствующих невральных и экстраневральных расстройствах.
Последствия спинномозговых повреждений зависят от уровня миелоцеле, который в более 80% случаев располагается в пояснично-крестцовой области. У детей наблюдается различной степени вялость, арефлекторный паралич и сенсорный дефицит, который может быть асимметричным, особенно при высоком уровне повреждения. Картина обычно складывается из смешанной дисфункции верхних и нижних двигательных нейронов. Двигательный дефицит часто неравномерный. Уровень чувствительности более устойчив, позволяя точно определить верхнюю границу повреждения. Спустя месяцы и годы становятся заметны истощение, плохая микроциркуляция в коже и трофические изменения. Ретракция приводит к тугоподвижности и деформации, а недостаточная оссификация связана со срастающимися в неправильном положении переломами. Функции сфинктера и детрузора нарушаются во всех случаях.
При повреждениях выше уровня L3 наблюдается полная параплегия. Нижние конечности при этом совершенно вялые, но прямые. Сенсорный дефицит присутствует дистальнее дерматомов L3 или L4. Способность передвигаться после операции отсутствует. При низких поясничных повреждениях сохранены функции сгибателей, приводящих мышц бедра и разгибателей колена. Вывих бедра часто существует с рождения, и сгибательная контрактура тазобедренного сустава приводит к сгибанию конечностей в бедрах с выпрямлением колена. С соответствующим лечением часто возможно передвижение с помощью. При повреждениях верхних крестцовых корешков передвижение возможно при минимальной помощи или самостоятельно. Степень поражения стоп различна, но в большинстве случаев присутствует определенная деформация. При крестцовых повреждениях ниже S3 функция нижних конечностей сохранена на нормальном уровне, но с отсутствием чувствительности в области седла и дисфункция сфинктера.
Патология чувствительности может вызвать трофические изменения кожи, переломы с избыточным формированием мозоли или артропатии.
Двигательные и сенсорные расстройства регистрируются в первые недели жизни ребенка. В течение первых 48 часов часто отмечают быстрое ухудшение моторной функции, что, вероятно, обусловлено механической травмой невральной пластинки во время родового процесса и/или внешним воздействием. Наоборот, после закрытия дефекта может наблюдаться стремительное улучшение. Позднее послеоперационное ухудшение не относится к естественному течению миеломенингоцеле, а указывает на осложнения (Hunt, 1990). Среди них различают сдавление реконструированного арахноидального пространства, ограничение спинного мозга рубцами, гидросирингомиелию, сегментарные инфаркты мозга и сдавление мозга кистами или липомами. В таких случаях КТ или МРТ завершая радиальную миграцию поможет пролить свет на причину расстройства и найти эффективное лечение.
Дисфункция сфинктера проявляется в двух основных типах, часто сосуществующих (Fernandes et al., 1994). При поражении ниже уровня S3 мочевой пузырь и анальный сфинктер парализованы с утратой чувствительности в прямой кишке и нижних отделах мочевыводящих путей. У трети пациентов появляется подтекание: мочевой пузырь переполнен с отсутствием активности детрузора по данным цистометрии (Borzyskowsky, 1990), и моча без затруднений выделяется при ручном нажатии в надлобковой области. В наиболее многочисленной группе с повреждениями на более высоком уровне сокращения детрузора слабые и затрудненная дефекация в результате нарушенной координации между сокращениями наружного сфинктера и детрузора. Чувствительность мочевого пузыря непостоянна. Это приводит к высокому уровню внутрипузырного давления с образованием трабекул и в конечном счете к расширению верхних отделов мочевыводящих путей. Бактериурия наблюдается у 50% двухлетних детей; инфекция мочевыводящих путей приводит к хроническому пиелонефриту, что является самой частой причиной смертности и заболеваемости таких пациентов (Hunt и Whitaker, 1987, Borzyskowski, 1990).
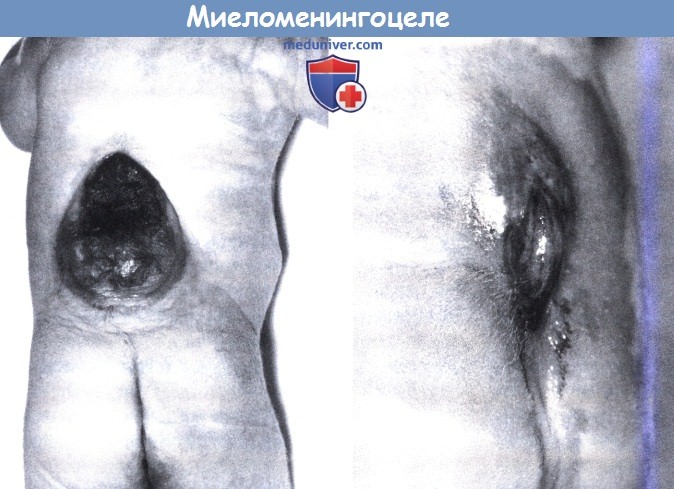
Миеломенингоцеле, орсолюмбальное повреждение, сопровождающееся параплегией и гидроцефалией.
(слева). Люмбосакральное повреждение, ясно указывающее на спинной мозг без нейруляции.
в) Гидроцефалия является основным осложнением миеломенингоцеле. Простая непрогрессирующая дилятация желудочков не создает больших трудностей. Прогрессирующая гидроцефалия нередко присутствует, несмотря на нормальную кривую роста окружности головы. Гидроцефалия с нормальным давлением может иметь место в некоторых случаях менингоцеле, и этим детям поможет шунтирование. По данным ультрасонографии, гидроцефалия присутствует при рождении в 85-95% случаев. Этого не наблюдается при крестцовых дефектах. Клинические проявления прогрессирующей гидроцефалии у пациентов с миелоцеле обычно не специфичны. Особые проявления, однако, могут быть результатом вовлечения нижних черепных нервов, возникающего, видимо, из-за сочетания аномалий ствола мозга и гидроцефалии.
г) Обструкция верхних дыхательных путей из-за паралича голосовой щели и центральное апноэ являются осложнениями мальформации Киари II и представляют основную причину смерти таких пациентов (Papasozomenos и Roessmann, 1981). Возможны и другие нарушения дыхания, в целом известные как центральная вентиляционная дисфункция (ЦВД). ЦВД включает помимо обструкции респираторных путей, периодическое учащение дыхания во сне (Ward et al., 1986а) и центральное апноэ (Oren et al., 1986). У некоторых детей снижена или отсутствует реакция на гипоксию и гиперкапнию (Ward et al., 1986b, Petersen et al., 1995). Частота ЦВД у пациентов, страдающих гидроцефалией колеблется от 5,7% (Hays et al., 1989) до 20% (Kirk et al., 2000). Первые симптомы появляются в течение одного месяца после рождения в двух третях случаев. Во всех случаях ЦВД присутствует гидроцефалия. Симптомы могут включать только стридор или стридор, сопровождающийся приступами цианоза и/или эпизодами апноэ. Прогноз неблагоприятен: 19 из 35 пациентов (Hays et al.) умерли до 30 месяца жизни. ЦВД может изменяться и исчезать. В некоторых случаях коррекция дисфункции шунта может облегчить симптомы ЦВД, если считать возможной причиной высокое внутричерепное давление со сдавлением ствола мозга (Hays et al., 1989). В некоторых случаях, вероятно, имеется более широкая картина дисфункции нижних черепных нервов, включая дисфагию, брадикардию, плохое самочувствие и слабость в верхних конечностях с возможностью разрешения при снижении внутричерепного давления.
Такие случаи младенческого стволового синдрома требуют неотложной помощи. Часто встречаются синдром ночного апноэ. Deray et al. (1995) обнаружили патологические изменения при полисомнографических исследованиях у 14 из 16 симптоматических пациентов. Поэтому исследования сна должны выполняться систематически. Тяжелые аномалии сопровождаются дополнительной патологией, такой как миеломаляция и тяжелый спинальный стеноз. У большинства бессимптомных пациентов имеются нормальные результаты исследования и хорошее самочувствие без хирургического лечения. Однако Petersen et al. (1995) заключили, что пневмограмма и испытание 10% С02 недостоверно прогнозируют развитие симптомов у новорожденных. Taylor et al. (1996) подчеркнули значение вызванных стволовых потенциалов, патологические значения которых имеют хорошую корреляцию с респираторными проблемами и неврологическими последствиями.
Лечение ЦВД может потребовать тонзилэктомии или трахеостомии отдельно или с хирургической декомпрессией задней ямки (Navarro et al., 2004). Однако практика в подобных случаях основана на несистематическом клиническом опыте и достичь первоклассного доказательного уровня не так просто (Tubbs и Oakes, 2004). Дисфагия в отдельности не требует хирургического лечения и имеет благоприятный прогноз.
Другим специфическим осложнением гидроцефалии в связи с миеломенингоцеле являются поздние признаки значительного и развивающегося сколиоза с прогрессирующим парезом. Такие неврологические проявления должны быть выявлены как можно раньше, так как проверка плохо функционирующего шунта может улучшить значительный дефицит (Hoffman et al., 1987).
Умственная отсталость обычно является следствием гидроцефалии, хотя могут играть роль и сопутствующие мальформации. Дата шунтирования, количество ревизий шунта и, особенно, инфекции ЦНС определяют конечный уровень интеллекта. Значительное ухудшение отмечается у инфицированных пациентов с шунтом, особенно при вентрикулите. Судороги часто возникают у пациентов с гидроцефалией в связи с миеломенингоцеле (Talwar et al., 1995).
д) Ведение spina bifida cystica. Ведение spina bifida cystica остается сложной проблемой до сих пор не имеющей удовлетворительного решения. Лучшей формой терапии, безусловно, является профилактика. Первичная профилактика с использованием витаминов и фолиевой кислоты эффективна и безопасна. Поливитаминные препараты, содержащие фолиевую кислоту, обязательны для беременных женщин, у которых рождались больные дети и, вероятно, показаны при всех беременностях (MRC Study Research Group, 1991). Пренатальный диагноз в настоящее время наиболее эффективен для предупреждения рождения больных детей; рекомендуемое скрининговое исследование материнской крови на а-ФП экономически нецелесообразно в регионах с низкой частотой spina bifida. Для больного плода профилактическое кесарево сечение не может считаться универсальным вариантом, предпочтительнее вагинальные роды.
Показания к восстановлению миеломенингоцеле изменялись с течением времени. В 1960-х годах за срочное восстановление выступали во всех случаях. В 70-х годах некоторые исследователи сообщали, что в случаях с неблагоприятным прогнозом — например, высокое поясничное повреждение со значительным кифозом, полной вялой параплегией и параличом мочевого пузыря, окружностью головы, по крайней мере, на 2 см больше 90-го центиля—показатели выживаемости хуже (Lorber, 1975). Неоперированные новорожденные имели двухлетний период выживаемости на уровне 0-4%. Многие клиницисты сегодня отказываются «выбирать» пациентов для операции за исключением, пожалуй, экстремальных случаев, таких как небольшие недоношенные, ослабленные новорожденные. Действительно, продолжительные проблемы бывают и у неоперированных детей, а некоторые дети с тяжелыми неблагоприятными критериями были способны прожить продуктивную жизнь. Фактически, операция обеспечивает сохранение функции, а не жизни, и в настоящее время выполняется в большинстве случаев. Оперативное вмешательство может быть выполнено в неонатальном периоде или отложено на три месяца. Дефект должен быть закрыт в стерильных условиях. В США начато перспективное исследование по сравнению эффективности раннего и позднего проведения операции (Lancet, 2004).
Пренатальное закрытие открытого дефекта было проведено в ряде случаев как попытка улучшения функциональных моторных и урологических исходов и уменьшения частоты и тяжести мальформации Киари II (Walsh и Adzyck, 2003, Kaufman, 2004, Tulipan, 2004, Zambelli et al., 2007). Эффективность не подтвердилась, кроме того, необходима полная оценка риска. Гидроцефалия, требующая шунтирования, встречается часто (Bruner et al., 2004). По данным одного исследования, риск осложнений при преждевременных родах не был значительно выше показателей в контрольной группе (Hamdan et al., 2004).
Методика хирургического закрытия дефекта также различается. Современные технологии обеспечивают лучшую подвижность спинного мозга относительно костей и кожных покровов. Закрытие дефекта облегчает уход за ребенком, уменьшает риск восходящей инфекции, способно улучшить двигательную и сенсорную функции. Вторичное ограничение подвижности спинного мозга возникает в 10% случаев с отсроченными симптомами, вызывающими необходимость дальнейших операций. Позднее закрытие проводится при больших дефектах (Ersahin и Yurtseven, 2004) или у маленьких ослабленных новорожденных.
Операция является первым шагом в ведении spina bifida (Kaufman, 2004). Возникающие у пациентов с менингоцеле проблемы таковы, что для успешного ведения необходимо привлечение детских хирургов, педиатров, урологов, физиотерапевтов и психологов (Kaufman, 2004). Нейрохирургическое обеспечение после восстановления примерно в 90% случаев включает шунтирование прогрессирующей гидроцефалии, а иногда и декомпрессию задней ямки в случаях сдавления спинного мозга в шейном отделе с затруднением дыхания и параличом нижних черепных нервов (см. выше).
Контрактурная деформация нижних конечностей требует проведения физиотерапии, фиксации в корсете или ортопедических операций, таких как мышечная трансплантация или суставной артродез. Необходимо тщательное наблюдение за тазобедренным суставом из-за частых дислокаций.
Урологические проблемы имеют первостепенную важность в контроле и лечении пациентов со spina bifida. Читатели могут обратиться к работам Borzyskowski (1990) и Snodgrass и Adams (2004). Основной задачей является профилактика инфекций мочевых путей и защита почек. Последнее особенно важно в случаях спастического, раздражительного пузыря с пузырно-мочеточниковым рефлюксом и может потребовать хирургического пособия. Регуляция работы мочевого пузыря остается проблемой. В ряде случаев эффективны соответствующее обучение и уход. Для остальных могут применяться различные методы. При существенном объеме мочевого пузыря и отсутствии пузырно-мочеточникового рефлюкса у небольшого числа детей может быть эффективным отжимание мочевого пузыря (Borzyskowski, 1990, Kothari et al., 1995). Медикаментозное лечение бромидом пропантелина или феноксибензамином малозначимо, и продолжительный дренаж мочевого пузыря оценивается по-разному. Трансуретральная резекция может выполнять небольшую роль в лечении.
При недержании важным мероприятием становиться периодическая катетеризация пузыря с учетом особенностей хода мочеиспускательного канала у мальчиков. Хорошие результаты достигаются различными методами хирургического отведения мочи (Chulamorkodt et al., 2004, Snodgrass и Adams, 2004). Основной причиной заболеваемости и смертности у пациентов со spina bifida являются инфекции и расширение верхних отделов мочевого тракта. Поэтому основой ведения больных становится раннее и регулярное наблюдение за мочевым трактом с выделением мочевых культур, внутривенными пиелограммами и цистограммами. Анальное недержание является меньшей проблемой и обычно успешно купируется клизмой и тренировками. Оценка уродинамики у детей с нейропатией мочевого пузыря (Rickwood, 1990) полезна для определения типа дисфункции мочевого пузыря и соответствующего ведения. Borzyskowski (1990) и Stephenson и Mundy (1990) дали подробный обзор методов консервативного и хирургического лечения.
Прогноз миеломенингоцеле остается тяжелым, даже при возможности продуктивной жизни в ряде случаев.
е) Менингоцеле. Менингоцеле — часть основной патологической картины миеломенингоцеле, но мезенхимальный дефект покрыт кожей, а спинной мозг без повреждений. Грыжевое выпячивание через затылочную кость и мышечный дефект ограничено мозговыми оболочками. В некоторых случаях корешки могут проникать в стенку менингоцеле или пересекать его полость. При операции хирургу необходимо скрупулезно оценивать любые «фиброзные» тяжи с помощью операционного микроскопа и электростимулятора.
Менингоцеле никогда не сопровождаются развитием гидроцефалии. Анатомическое распределение аналогично миеломенингоцеле. Менингоцеле может быть плоским или на ножке. В последних случаях твердая или паутинная мозговая оболочка имеют тенденцию к соединению в шейке мешка, а развитие адгезии может частично нарушить нормальное сообщение между мешком и субарахноидальным пространством (Friede, 1989). При поражениях на ножке костный дефект обычно ограничен одним или двумя задними сводами.
Клинически менингоцеле отмечается выпячиванием кожи над флюктуирующими массами. Нередко изменение кожного покрова с обширными ангиомами. Неврологическое обследование отрицательное. Менингоцеле не требует неотложного восстановления, и обычно предпочтительнее плановое закрытие позднее, в детском возрасте.
Вентральные менингоцеле встречаются наиболее редко. В основном они локализуются в области крестца и могут достигать значительных размеров из-за частичной аплазии крестцового отдела. Передние крестцовые менингоцеле могут наследоваться по доминантному типу (Gardner и Albright, 2006). Обычно они протекают незаметно на протяжении детского возраста, хотя способны провоцировать ректальную компрессию и хронические запоры (Ashley и Wright, 2006). Синдром Куррарино (Emans et al., 2005) включает триаду из крестцовой агенезии, аноректальной мальформации и пресакральной менингоцеле и/или другие пре-сакральные образования, такие как тератома. Связь с ЦСЖ часто сопровождается неврологическими осложнениями (Cretolle et al., 2006). Это связано с доминирующей мутацией в homeogene HLXB9 (Merello et al., 2006). Синдром Куррарино — одна из форм синдрома каудальной регрессии (Singh et al., 2005) с варьирующим выражением в различных аномалиях дистальных сегментов спинного мозга и нижнего отдела позвоночника с неврологическими и сфинктерными нарушениями. Около 1% случаев связано с материнским диабетом (Zaw и Stone, 2002). Наиболее тяжелой формой является сиреномиелия. Наиболее частое проявление — частичная или тотальная агенезия крестца. Wilmshurst et al. (1999) подробно описали проблемы ведения на основе обзора клинических и рентгенологических описаний 22 случаев.
Латеральные менингоцеле являются грыжевыми выпячиваниями патологически тонкой твердой мозговой оболочки через увеличенное латеральное отверстие. Они проявляются нейрофиброматозом совместно с зазубриванием тел позвонков.
ж) Spina bifida occulta (скрытый спинальный дизрафизм). Термин «spina bifida occulta» применим в случаях спинального дизрафизма, при котором нет грыжевого выпячивания нервных структур или оболочек через мезенхимальный дефект. Это определение включает синдром расщепления хорды, дорсальные дермальные синусы, фибролипомы конского хвоста и диастематомиелию. Липомы и липомиеломенингоцеле не совсем отвечают критериям скрытого дизрафизма, потому что вызывают определенное выбухание кожи и повреждение спинного мозга, очень близкое к классическому миеломенингоцеле. Однако они будут описаны в этом разделе, так как их клиническая симптоматика не сильно отличается от других форм скрытого дизрафизма.
Клинические проявления spina bifida occulta могут быть разными. Липомы, липоменингоцеле, диастематомиелия и синдром фиксированного спинного мозга имеют много общих признаков, и клинические проявления не требуют отдельного описания. Образования кожи, такие как ангиомы, ямочки, полипы, кисты или патологические участки оволосения чрезвычайно распространены и являются диагностическим признаком (Henriques et al., 2005). Во всех случаях обязательно определение границ и природы повреждения при детальном обследовании. Целесообразны рентгенография в обычной проекции, КТ с/без метризамидом и МРТ. Оптимально проведение МРТ, обеспечивающего возможность исследования в нескольких плоскостях. Ультрасонография позвоночника и спинного мозга эффективны, хотя и менее точны (Robinson et al., 2005). Гидроцефалия не является признаком скрытого спинального дизрафизма. Мальформация Киари II сопровождается дизрафизмом лишь в редких случаях (Tubbs и Oakes, 2004).
з) Синдром расщепления хорды. Этот синдром является следствием расщепления хорды во время эмбриогенеза с устойчивой связью между кишкой и дорсальной кожей. Обычно присутствует лишь частичная связь между кишкой и позвоночником или спинным мозгом. Тотальная форма (дорсальная кишечная фистула) встречается редко. Интраспинальные кишечные кисты в основном находят в верхнем грудном отделе. Они выстланы кишечным эпителием, могут вызывать компрессию спинного мозга и должны лечиться хирургическим способом (Rebhandl et al., 1998).