Сочетанные и осложненные спинальные мышечные атрофии (СМА) у детей
Спинальная мышечная атрофия (СМА) встречается в сочетании с поражением структур ЦНС при нескольких редких полисистемных нарушениях, из которых наиболее важен боковой амиотрофический склероз (БАС), являющийся главным образом спорадической болезнью взрослых и клинически характеризующийся сочетанием поражения нижних мотонейронов с пирамидными знаками. Приблизительно 10% случаев детерминированы генетически и вызваны, по меньшей мере, восемью различными мутациями.
Боковой амиотрофический склероз (БАС) редко встречается у молодых людей (ювенильный боковой амиотрофический склероз (БАС)), вместе с тем было описано несколько мутаций и клинических форм.
а) Ювенильный боковой амиотрофический склероз. Клинические проявления ювенильного бокового амиотрофического склероза (БАС) вариабельны. Большинство случаев очевидно относится к БАС2 и представляет собой хроническое заболевание с амиотрофией в верхних конечностях, а иногда также с бульбарной амиотрофией с двусторонними пирамидными знаками, сильно напоминая боковой амиотрофический склероз взрослого типа, но с относительно доброкачественным медленным течением и семейным (аутосомно-рецессивным) характером.
Истинные случаи БАС трудно отделить от других состояний с хронической амиотрофией и некоторых форм семейной спастической параплегии. Пациенты, описанные в Тунисе Ben Hamida et al. (1990), относились к трем подгруппам. Самая многочисленная группа состояла из клинически типичных случаев, в которых позднее Hentati et al. (1994) выявили связь с мутацией гена на участке хромосомы 2q33, кодирующего белок альсин (англ, alsin от ALS — Amyotrophic Lateral Sclerosis — боковой амиотрофический склероз).
Две другие группы включали случаи спастической параплегии, связанной с перонеальной атрофией или со спастическим и псевдобульбарным синдромом, и расцениваются как форма наследственной спастической параплегии, одна из которых связана с мутацией в гене альсина (Gros-Louis et al., 2003).
Наследственный восходящий спастический паралич с началом в раннем детстве (НВСПР, англ. IAHSP) (Eymard-Pierre et al., 2002), клинически отличается от БАС, но также происходит из-за мутации в гене альсина. Lesca et al. (2003) сообщили о 16 случаях НВСПР в 11 семьях из Европы и Северной Африки. Они возникали в первые два года жизни, начинаясь восходящей спастической параплегией, с вовлечением верхних конечностей в конце первого десятилетия. Затем — медленное прогрессирование на протяжении второго десятилетия, с результатом в виде тетраплегии, анартрии, дисфагии и замедленных движений глаз.
Интеллект оставался нормальным, и пациенты выживали в течение длительного времени. Признаки поражения периферических мотонейронов отсутствовали. Методы нейровизуализации показали двустороннее усиление сигнала в заднем бедре внутренней капсулы и определенную степень корковой атрофии у самых старших пациентов. Мутация гена альсина была найдена в четырех из 10 изученных семей, но была исключена в других случаях. Таким образом, синдром генетически гетерогенен. Несмотря на наличие мутации в гене альсина, он явно отличается от БАС, хотя нозологическая ситуация остается неясной (Eymard-Pierre et al., 2006).
Другие типы бокового амиотрофического склероза (БАС) с ранним началом очень редки. Боковой амиотрофический склероз (БАС) 4 похож на чисто моторную невропатию с некоторыми пирамидными компонентами и без бульбарных симптомов и связывается с 9q34 (De Jonghe et al., 2002). Боковой амиотрофический склероз (БАС) 5 связывается с участком хромосомы 15q15.1 — 15q21.1 (Hentati et al., 1994) с клиническими симптомами взрослого БАС, но более доброкачественным течением.
Семейное дегенеративное заболевание клеток передних рогов и пирамидных трактов, часто в сочетании с паркинсонизмом, деменцией и течением, приводящим к летальному исходу, все еще встречается среди народа чаморро живущего на острове Гуам в Тихом океане, хотя его частота резко уменьшилась. Причина остается неизвестной (Galasko et al., 2002).
Спинальная мышечная атрофия (СМА) может также встречаться в сочетании с поражением задних столбов спинного мозга (Engel et al. 1959), и известны более комплексные случаи, включая пирамидное, спиноцеребеллярное и таламическое поражение (Grunnet и Donaldson, 1985).
б) Первичный боковой склероз. Это состояние встречается исключительно у детей (Grunnett et al., 1989), проявляясь прогрессирующим спастическим и бульбарным синдромами (Lerman-Sagie et al., 1996). О семейных случаях, связанных с псевдобульбарным синдромом и парезом взора, сообщали Gascon et al. (1995). В некоторых случаях была найдена мутация в гене альсина на участке хромосомы 2q33 (Yang et al., 2001).
в) Агенезия мозолистого тела с нейронопатией (синдром Андерманна). Синдром, проявляющийся агенезией мозолистого тела, болезнью клеток передних рогов, смешанной сенсорной и моторной невропатией и лицевым дизморфизмом, встречается в Квебеке и наследуется по аутосомно-рецессивному типу (Larbrisseau et al., 1984, Hauser et al., 1993). Ген идентифицирован (Howard et al., 2002). Умеренная задержка интеллектуального развития также симптоматична. Течение является медленно прогрессирующим, но продолжительность жизни представляется нормальной.
г) Другие осложненные спинальные мышечные атрофии (СМА). Взрослая форма дефицита гексозаминидазы А может встречаться в детстве. Она регулярно проявляется поражением клеток передних рогов с медленно прогрессирующим течением, начинающимся в половине случаев в возрасте до 10 лет. Часто наблюдаются сопутствующие дистония и пирамидные знаки (Specola et al., 1990), но иногда выявляется и изолированное поражение клеток передних рогов (Johnson et al., 1982).
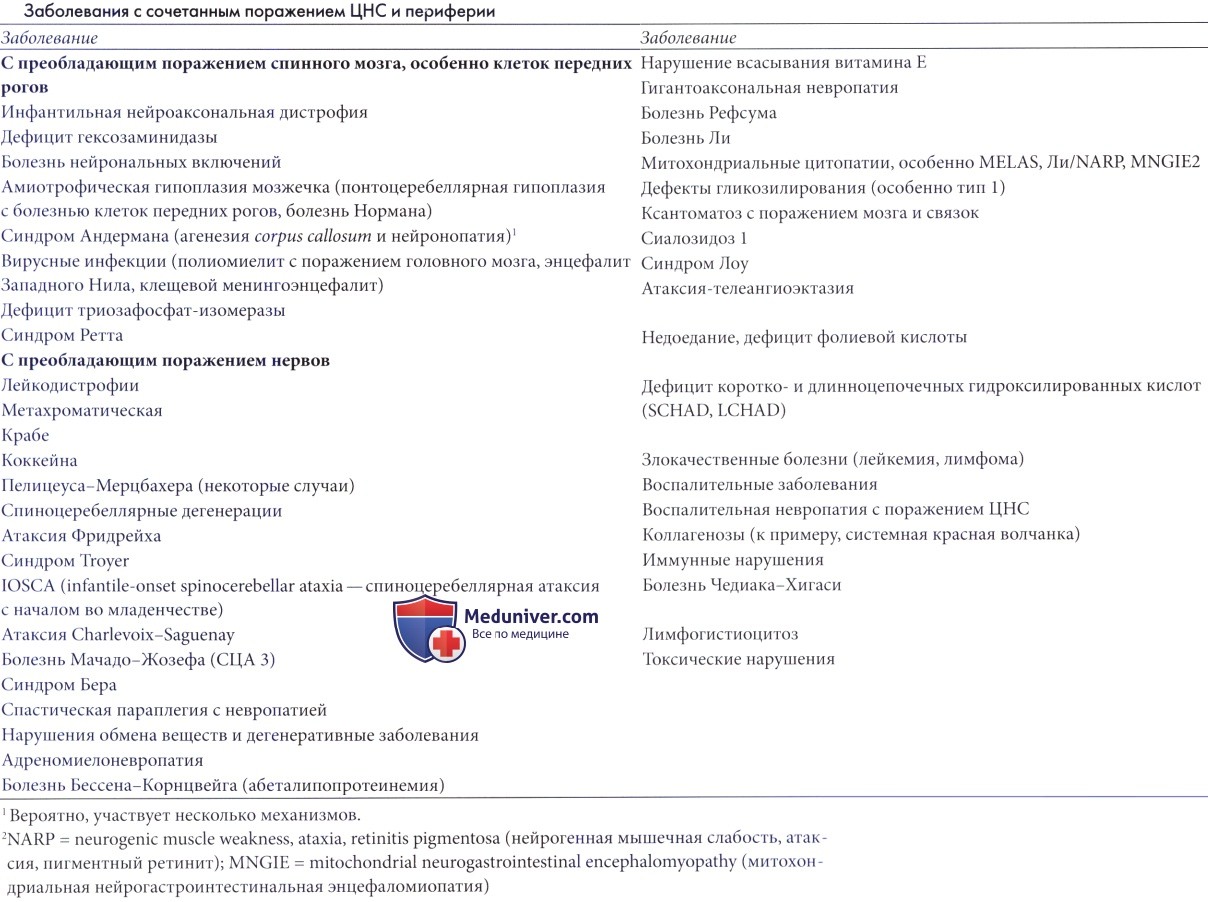
д) Спинальные мышечные атрофии (СМА) с пигментным ретинитом. Были описаны редкие случаи семейной прогрессирующей атрофии рук, плеч, шеи и груди с потерей сухожильных рефлексов и ассоциированной тугоухостью, типичной ретинопатией с костным тельцем и олигофренией (Walsh и Hoyt, 1969). Вероятно, присутствовал спастический компонент. Взаимозависимость пигментного ретинита и мышечной слабости гораздо чаще является следствием митохондриальной болезни, и тогда слабость обычно имеет миогенное происхождение. Несколько других состояний, проявляющихся поражением ЦНС и периферии, перечислены в таблице ниже.
В нее включены и заболевания, поражающие аксоны в большей степени, чем передние рога, так как связанные с ними клинические проблемы очень сходны с таковыми при поражении передних рогов.
е) Приобретенные болезни мотонейрона. Приобретенные болезни мотонейрона имеют главным образом вирусное происхождение; роль токсических, сосудистых или других факторов окружающей среды в этиологии бокового амиотрофического склероза и других родственных синдромов обсуждена Wicklund (2005).
Большинство приобретенных вирусных заболеваний клеток передних рогов протекают остро. В основном они представлены острым полиомиелитом и похожими заболеваниями, вызванными энтеровирусами, не относящимися к полиовирусам.
Острый полиомиелит является, безусловно, наиболее частой причиной приобретенного заболевания клеток передних рогов. Он, к сожалению, по-прежнему распространен в некоторых развивающихся странах. В развитых странах встречаются только редкие случаи у непривитых детей. Сообщалось о казуистических случаях паралитического полиомиелита из-за вируса ослабленной живой вакцины у реципиентов вакцины или при контактах, главным образом у детей с иммунодефицитом. Полиомиелит обсуждался в главе 10. Сообщалось о полиомиелитоподобных заболеваниях из-за других энтеровирусов, включая вирусы групп ЕСНОвирусы и коксаки-вирус.
Энцефалит Западного Нила — передаваемая клещами флавивирусная инфекция, был ответственен за эпидемии менингоэнцефалита в нескольких Американских штатах. Он также может вызвать полиомиелитоподобное заболевание с начальной лихорадкой и менингеальными знаками, с плеоцитозом в ЦСЖ, через несколько дней за ним следует фокальный асимметричный вялый паралич, который может быть более или менее значительным (Jeha et al., 2003). Электрофизиологические особенности неотличимы от таковых при паралитическом полиомиелите (Al-Shekhlee и Katirji, 2004), и МРТ в Т2-режиме может выявить повышение плотности клеток передних рогов (Jeha et al., 2003).
Подобная болезнь также может быть следствием флавивирусной инфекции японского энцефалита (Агуа, 1998). С развитием авиапутешествий подобные случаи, вероятно, будут наблюдаться и в Европе. Сообщалось о редких случаях полиомиелитоподобной болезни, вызванной вирусом европейского клещевого энцефалита (Aendekerk et al., 1996).
Синдром Хопкинса, который заключается в остром полиомиелитоподобном параличе после приступа астмы, может иметь вирусное происхождение (Arita et al., 1995).