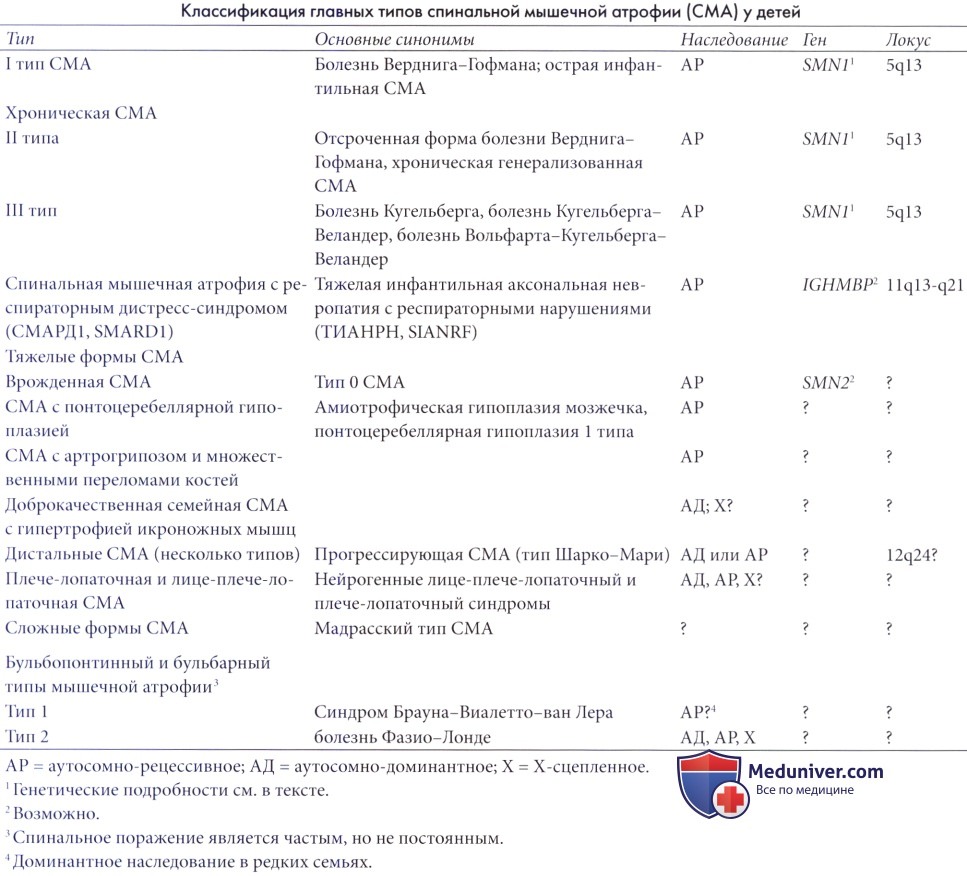
Классификация спинальной мышечной атрофии (СМА) у детей
После практически полного исчезновения острого полиомиелита в развитых странах на первое место среди заболеваний клеток передних рогов вышли хронические дегенеративные заболевания. В некоторых развивающихся странах, однако, острый полиомиелит остается причиной поражения моторных нейронов спинного мозга.
Спинальные мышечные атрофии составляют гетерогенную группу заболеваний, характеризующихся прогрессирующей дегенерацией клеток передних рогов спинного мозга и часто клеток моторных ядер в стволе мозга. В совокупности они представляют самое частое дегенеративное заболевание ЦНС и второе по частоте после муковисцидоза смертельное генетическое заболевание детского возраста. В основном это генетические болезни с различными способами наследования. Начало может быть в любом возрасте от младенчества, или даже пренатального периода, до взрослой жизни. Отвечающие за нейрональную дегенерацию и смерть механизмы пока еще не совсем ясны.
Вовлечение моторных нейронов может быть диффузным или ограниченным определенными мышечными группами. Чаще всего мышечная слабость и атрофия затрагивают проксимальную часть конечностей, но известны дистальная, плече-лопаточная, лице-плечелопаточная и сегментарная формы, и топография вовлечения и способ наследования важны для диагноза.
Недавно были представлены надежные доказательства, что эти нарушения могут также затронуть аксоны и множество нейронов за пределами передних рогов.
Классификация спинальной мышечной атрофии (СМА) затруднительна, ни одна из предлагавшихся систем не была удовлетворительной. Было достигнуто соглашение по классифицированию трех типов наиболее распространенных форм. Тип I, или болезнь Верднига-Гофмана, обозначает легко распознаваемую острую форму с ранним началом.
Хронические формы разделены на тип II с более поздним началом на первом году жизни, а топография парезов диффузна с результатом в виде деформаций и серьезного снижения функциональности, и более умеренный тип III, также известный как болезнь Кугельберга, Кугельберга-Веландер или Вольфарта-Кугельберга с более поздним началом и преимущественно проксимальным поражением (Реагп, 1980; Emery, 1994). Dubowitz (1999) предложил дополнение в виде очень тяжелой формы с пренатальным началом (тип 0), которая заканчивается внутриутробной гибелью или краткосрочным выживанием лишь при неотложной респираторной поддержке. Фактически доказано, что спинальные мышечные атрофии (СМА) могут формировать фенотипический континуум с промежуточными формами между всеми тремя классическими типами (Iannaccone, 1998; Dubowitz, 1999). Эта классификация вытеснила предыдущие схемы.
Классические спинальные мышечные атрофии (СМА) отвечают следующим критериям (Munsat и Davies, 1992): симметричная слабость мышц туловища и конечностей, более заметная проксимально, чем дистально, и преобладающая в нижних конечностях; наличие фасцикуляций языка и/или тремора пальцев, известного также как миниполиоклонус, характерный для поражения клеток передних рогов; электромиограмма и биоптат с признаками типичных нейрогенных нарушений. Напротив, критериями исключения являются: нарушения чувствительности, наличие патологии ЦНС, обширный артрогрипоз, более чем десятикратное повышение креатинкиназы относительно максимального нормального уровня, и снижение скорости проводимости нерва более 70%. Тем не менее, некоторые критерии необходимо переоценить с точки зрения данных последних исследований, указывающих на более диффузное поражение.
Было продемонстрировано, что три классических типа являются аллельными и связаны с маркерами хромосомного локуса 5q13 (Brzustowicz et al., 1990; Melki et al., 1990a, b), в этом локусе был идентифицирован ген SMN (Survival Motor Neurons — выживание мотонейронов), кодирующий ранее неизвестный белок из 294 аминокислот (Melki et al., 1994; Lefebvre et al., 1995). Делеция экзона 7 в этом гене найдена в 85-95% всех типов СМА (Rodrigues et al., 1996). Гомозиготная делеция гена SMN была выявлена у 93% и разрыв гена — у 5,6% пациентов с тяжелой СМА.
В целом тяжелые формы связаны с большими делециями, которые также удаляют смежные гены (особенно ген DNAI), но делеция экзона 7 достаточна, чтобы вызвать тяжелые формы. В редких случаях ген не отсутствует ни полностью, ни частично, но присутствует точечная мутация в экзоне 7, поддерживая предположение, что ген SMN — и особенно экзон 7 — является ответственным за фенотип СМА. Однако ген SMN присутствует в двух копиях (теломерный или SMN1 и центромерный или SMN2), и SMN2 может кодировать некий защитный белок и выполнять компенсаторную роль и мог бы «спасти» функцию SMN1, даже при том, что это «спасение» не может быть достаточным в большинстве случаев. В результате число копий гена SMN2 может влиять на тяжесть клинической картины.
Были идентифицированы редкие случаи здоровых носителей с гомологичной делецией гена SMN 1 (Cobben et al., 1995; Prior et al., 2004). В некоторых таких случаях рассматривалась компенсаторная роль гена SMN2, таким образом, определенная неуверенность сохраняется даже при однозначном признании основной роли гена SMN1. Некоторые исследователи (DiDonato et al., 1997) предположили, что причиной II и III типов спинальной мышечной атрофии (СМА) часто является конверсия (т.е. обмен последовательностями ДНК между центральным и теломерными генами SMN с обретением теломерным локусом нуклеотидов, в норме ассоциированных с центромерным локусом, который неспособен кодировать экзон 7, но может кодировать экзон 8). Такая конверсия, неотличимая при помощи обычных методов от делеции экзона 7, представляется в большей степени связанной с хроническими, обычно доброкачественными спинальными мышечными атрофиями (СМА), в то время как истинная делеция экзона 7 вызывает I тип.
Тем не менее, во многих случаях более вероятна делеция, а не конверсия, и множественные копии гена SMN2 в большинстве случаев не способны на последовательное «спасение» делеции SMN1 (Diep Tran et al., 2001; Harada et al., 2002).
He все случаи спинальной мышечной атрофии (СМА) ассоциированы с делецией генов SMN. Некоторые менее частые формы спинальной мышечной атрофии (СМА) не сцеплены с этим геном или с хромосомой 5. Они включают синдром СМАРД1(SMARD1) с тяжелым диафрагмальным параличом, случаи понтоцеребеллярной атрофии с спинальной мышечной атрофией (СМА), некоторые из случаев СМА, ассоциированной с артрогрипозом и врожденными переломами, и редкие случаи СМА с миоклонической эпилепсией (Haliloglu et al., 2002; Striano et al., 2004). Однако отдельные случаи с ассоциированным врожденным пороком сердца и с артрогрипозом и врожденными переломами демонстрируют такую же делецию экзона 7, как и классический тип (Burglen et al., 1996; Melki и Munnich, 1996; Garcia-Cabezas et al., 2004).