Острая инфантильная спинальная мышечная атрофия (болезнь Верднига-Гофмана, СМА первого типа)
Частота возникновения спинальной мышечной атрофии (СМА) I типа составляет 1 на 20 000 живорожденных, частота носительства гена 1 на 60-80 (Pearn, 1980). Она ассоциирована с делецией экзона 7 гена SMN более, чем в 95% случаев.
Делеция в гене NAIP (neuronal apoptosis inhibitory protein — белок, подавляющий нейрональный апоптоз) также найдена в 50-60% случаев I типа СМА (Rodrigues et al., 1996), и представляется, что большая делеция в области 5ql3 коррелирует с тяжелым типом СМА (Spiegel et al., 1996). Тем не менее, 27% случаев I типа СМА имеют только делецию гена SMN. Делеция в экзоне 3 была найдена у некоторых пациентов без делеции экзона 7 (Cobben et al., 1995), являясь результатом сдвига рамки и преждевременного терминирующего кодона.
а) Патоморфология. Основой патологии является бросающаяся в глаза гибель клеток передних рогов. Сохранившиеся мотонейроны участвуют в процессе дегенерации с хроматолизом и возможным фагоцитозом сателлитными клетками. Возможна поразительная сохранность нейронов в шейном отделе (Kuzuhara и Chou, 1981). Зоны глиоза наблюдались в передних корешках (Chou и Nonaka, 1978) и в сморщенных задних корешках. Значение этой находки остается неясным. Признаки вовлечения периферического нерва с потерей больших миелинизированных аксонов (Chien и Nonaka, 1989) скорее согласуются с валлеровской дегенерацией, чем указывают на признак процесса «отмирания».
Но в некоторых случаях имеет место более обширное сенсорное поражение (Anagnostou et al., 2005). Сенсорная невропатия была подтверждена в систематических исследованиях биоптатов икроножного нерва и пониженной скоростью сенсорной проводимости в 6 из 7 случаев СМА I типа, но не II или III типа, выявленной при молекулярных исследованиях (Rudnik-Schoneborn et al., 2003b). Супраспинальные очаги поражения равномерно представлены в моторных ядрах ствола мозга, особенно в ядре п. hypoglossus, nucleus ambiguus и ядре п. facialis. Часто встречается и вовлечение таламуса. Такие поражения могут быть ответственными за патологические изменения сенсорных вызванных потенциалов, включая соматосенсорные и зрительные потенциалы, о чем сообщают Cheliout-Heraut et al. (2003).
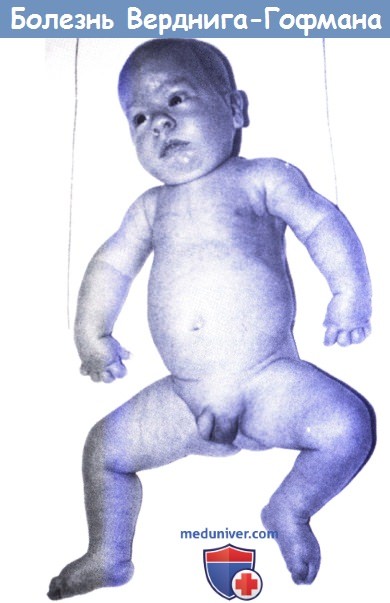
Болезнь Верднига-Гофмана (спинальная мышечная атрофия I типа).
Обратите внимание на суженную из-за паралича межреберных мышц грудную клетку, сгибательное положение пальцев, ретрогнатизм и настороженный вид.
б) Клинические признаки. Клинические признаки начала болезни характерны, позволяя почти сразу поставить диагноз за исключением случаев очень раннего начала. Приблизительно в 30% случаев начало относится к пренатальному периоду, и младенец рождается со слабостью проксимальных отделов конечностей и арефлексией. Слабость распространяется быстро, и через несколько недель отмечается тетраплегия с некоторой сохранностью движений в дистальных отделах, особенно в верхних конечностях. Паралич симметричен и также вовлекает мышцы, связанные с осевым скелетом, особенно шеи.
Паралич межреберных мышц — ключевой признак состояния. Он приводит к характерной деформации грудной клетки, которая распластана и остается неподвижной или, как это ни парадоксально, уменьшается в окружности при вдохе, тогда как живот выпячивается, напоминая движение качелей. Дыхательные движения выполняются почти исключительно диафрагмой, которая сохранна до поздних стадий болезни. Ретрогнатизм, иногда сопровождающийся фасцикуляцией мышц подбородка, является постоянным и, вместе с неизмененными движениями глаз и живым взглядом, завершает характерную картину внешнего вида пациентов.
Глубокие сухожильные рефлексы утрачены. Фасцикуляции языка часто присутствуют, но их трудно отличить от частых дрожательных движений языка у нормальных детей. Отсутствуют признаки сенсорного дефицита, пирамидных знаков и нарушений работы сфинктера. Интеллект сохранен, и младенцы обычно описываются как очень привлекательные.
Нарушения в большинстве случаев являются быстро прогрессирующими, особенно при пренатальных формах и в острых случаях раннего начала. Смерть наступает в течение первых 18 месяцев жизни в 80% случаев и является следствием дыхательной недостаточности, развитие которой часто ускоряют и утяжеляют интеркуррентные респираторные инфекции. У многих младенцев возникают проблемы с глотанием, вызывающие необходимость кормления через зонд. Пациенты с дебютом болезни в неонатальном периоде часто умирают до достижения возраста трех месяцев. Несколько сообщений (Russman et al., 1992; lannacone et al., 1993) указывают на менее мрачную перспективу с намного более высокими коэффициентами выживаемости и лучшей функциональностью, Chung и Wong (2004) также описали значительное улучшение показателей исходов у детей-китайцев. Однако не исключена определенная степень отбора случаев, и прогноз острой инфантильной СМА остается довольно плохим.
Тип 0 спинальной мышечной атрофии (СМА) (Dubowitz, 1999) обозначает самую тяжелую степень болезни с пренатальным началом, приводящую к смерти или неспособности начать и поддерживать дыхание с момента рождения. Эта форма требует неотложной механической респираторной поддержки, и внимания к значительным трудностям с глотанием. Похожие случаи описывают MacLeod et al. (1999). Даже в пределах этой формы степень тяжести может варьировать, подтверждая, таким образом, концепцию континуума тяжести заболевания.
Болезнь Верднига-Гофмана.
Биоптат мышцы, демонстрирующий пучковую (фасцикулярную) атрофию, типичную для поражения клеток передних рогов.
Обратите внимание, что атрофированные волокна сохраняют округлый контур.
Он необычен при спинальной мышечной атрофии с более поздним началом, при которой, как правило, наблюдаются угловатые атрофированные волокна.
в) Диагноз. Диагноз спинальной мышечной атрофии (СМА) I может быть подтвержден электромиографическим исследованием, которое показывает неврогенные изменения со снижением паттерна активности во время максимального усилия, увеличением продолжительности и амплитуды отдельных потенциалов моторной единицы, и увеличением сферы распространения полифазных потенциалов. Спонтанная активность в форме ритмической импульсации двигательных единиц присутствует в 69% случаев, и фибрилляции и положительные острые волны выявляются в 35% (Hausmanowa-Petrusewicz и Karwanska, 1986), но фасцикуляции редко видны у маленьких детей. Значительные потенциалы вызываются расширением зоны регенерации моторных единиц, принятой сохранившимися клетками передних рогов. Полифазные потенциалы распространены и, вероятно, являются результатом наличия в мышцах многочисленных групп мелких волокон без признаков реиннервации и менее плотно упакованных волокон в пределах двигательной единицы.
Скорость проведения по нерву нормальная или немного замедленная у большинства пациентов, но может наблюдаться более заметное снижение, особенно у тяжело пораженных пациентов (Imai et al., 1990).
Уровень креатинкиназы сыворотки обычно нормальный, хотя может быть несколько повышен у младенцев с быстро прогрессирующей формой.
Биопсия мышцы демонстрирует пучки маленьких округленных волокон, которые относятся как к I, так и ко II типу. Гипертрофированные волокна разбросаны среди атрофических пучков и принадлежат к типу I. Нормальный рисунок типа шахматной доски заменяется вариантом группировки больших количеств волокон одного типа. Биопсия мышцы не является необходимой для диагноза СМА, когда клинические и ЭМГ-данные характерны, и подтверждение оптимально при генетическом тестировании. Действительно, биопсию может быть трудно интерпретировать в начальной стадии заболевания или из-за проблем, связанных с препаратом, в то время как клинический и электромиографический диагноз не вызывает сложностей при интерпретации клинических признаков.
Диагностические тесты, основанные на анализе ДНК, в настоящее время заменили прежние диагностические методы. Они также эффективны для пренатальной диагностики (Stewart et al., 1998; Milunsky и Cheney, 1999) и имеют высокий уровень надежности благодаря отсутствию контаминации материнской ДНК. Jedrzejowska et al. (2005) смогли поставить диагноз в 263 из 266 случаев (96,6%), исследованных путем выявления экзона 7 стандартной ПЦР и количественной ПЦР в реальном времени для более сложных случаев. В настоящее время возможен достоверный предимплантационный диагноз (Malcov et al., 2004; Burlet et al., 2005).
Дифференциальный диагноз спинальной мышечной атрофии (СМА) типа 1 обычно не представляет сложности даже с учетом множества причин для гипотонии в грудном возрасте. Врожденные миопатии и врожденные мышечные дистрофии могут представлять сходную картину с отсутствием сухожильных рефлексов. Вовлечение дыхательной системы в подобных случаях отличается переднезадним выравниванием грудной клетки в противоположность боковому сжатию при СМА. Обычно присутствует поражение лица, а неврогенный ЭМГ-паттерн не наблюдается. Уровень креатинкиназы повышен. Обязательно нужно учитывать миастению как поддающееся лечению состояние, но клиническая картина отличается преобладающим в большинстве случаев вовлечением лица и глаз. Поперечное рассечение спинного мозга и врожденные спинальные опухоли в некоторых случаях могут демонстрировать поверхностное сходство, особенно в отношении вовлечения дыхательных мышц и деформации грудной клетки. Поэтому важно систематическое исследование чувствительности в нижней части тела и малых пирамидных знаков.
Инфантильный гликогенов типа 2 (болезнь Помпе), вызывающий подобное обширное вовлечение мышц, всегда связан с поражением сердца и, часто, с симптомами со стороны ЦНС. Описаны редкие случаи врожденной или ранней невропатии (Korinthenberg et al, 1997). У таких пациентов отмечается снижение нервной проводимости и возможны признаки сенсорных нарушений. В ЦСЖ может быть повышен белок (глава 20). Редкое митохондриальное заболевание с дефицитом цитохрома с вызывает тяжелую гипотонию с дыхательной недостаточностью в период новорожденности. Могут присутствовать почечные тубулярные симптомы и аномалии ЦНС (DiMauro et al., 1985). Необычная форма этой болезни возникает из-за преходящего дефицита цитохрома с, и выздоровление связано с отсроченным созреванием фермента (Zeviani et al., 1987). Необычные случаи гликогеноза IV типа могут давать сходную картину (Тау et al, 2004).
д) Лечение. Эффективное лечение еще недоступно. Показано интенсивное лечение респираторных инфекций. Механическая вентиляция легких вызывает расширение и рост легких и может продлить выживание пораженных младенцев и облегчить страдания родителей, но интенсивность терапии, и особенно показания к длительной механической вентиляции чрезвычайно сложно оценить с позиций гуманизма и этики (Gilgoff et al., 1989; Gordon, 1991).