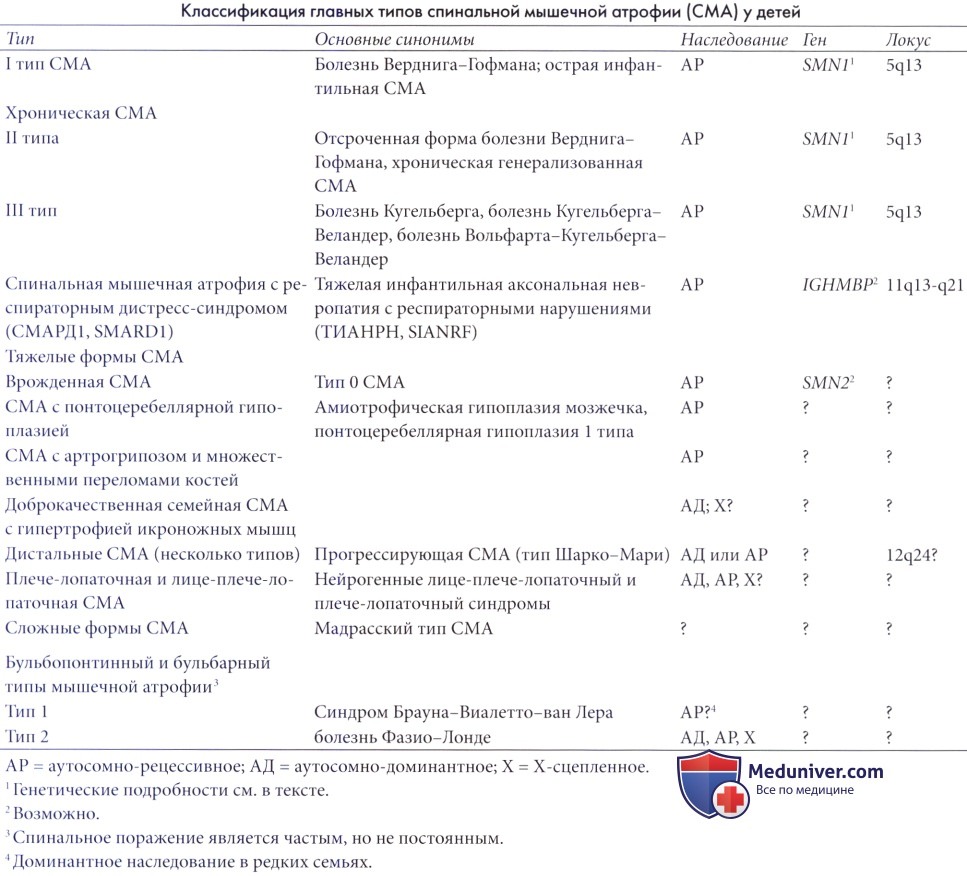
Редкие и нетрадиционные формы спинальной мышечной атрофии (СМА)
а) Доминантные диффузные (или проксимальные) формы хронических СМА. Доминантные формы хронических СМА редки в детском возрасте (Emery et al., 1976а, b; Pearn et al., 1978; Boylan и Cornblath, 1992). Характерно медленно прогрессирующее течение. Они представляют меньше чем 2% случаев в детстве, но до 30% взрослых случаев (Pearn, 1978) и также известны у взрослых как IV тип СМА. Возможно наличие двух различных генов: формы с ранним дебютом начинаются между рождением и восьмью годами без выраженной избирательности в отношении проксимальных мышц; формы с поздним дебютом наблюдались у молодых взрослых, и имеют явную проксимальную избирательность.
Недавно сообщалось о нескольких семьях с СМА с ранним дебютом, поражающей преимущественно нижние конечности с деформацией стоп и тяжелым нарушением способности передвигаться (Mercuri et al„ 2004b). Установлено, что клинически сходные случаи картированы на участке хромосомы 12q23-q24 (van der Vleuten et al., 1998). Некоторые другие случаи хронических СМА могут представлять собой негенетические фенокопии или новые доминантные мутации (Pearn, 1980).
б) Сцепленная с полом СМА. Редкие случаи сцепленной с полом СМА описаны у взрослых (Paulson et al., 1980) и детей (Skreet al., 1978). В семье, описанной Skre et al., у некоторых пациентов был диффузный паттерн поражения, а у других — лопаточно-перонеальный паттерн. Эти Х-сцепленные формы могут отличаться от проксимальной формы взрослой проксимальной нейронопатии или болезни Кеннеди (Guidetti et al., 1986), которая проявляется поражением бульбарных мышц, гинекомастией и частой асимметрией поражения в дополнение к проксимальной СМА.
Ген локализован в проксимальной части длинного плеча Х-хромосомы и включает CAG-повторы, экспансия которых связана с клиническим заболеванием. Ген кодирует андрогенный рецептор клеточной поверхности (Doyu et al, 1992).
в) Дистальная спинальная мышечная атрофия (СМА). Проявления этой формы сходны с таковыми при перонеальной мышечной атрофии. В большой серии Harding и Thomas (1980) это составляло 13% из 262 случаев. Вовлечены, по крайней мере, семь генов (Dierick et al. 2008), но нет никакой связи с 5 хромосомой. Доминантное наследование преобладало, но известны также и рецессивные формы (Harding и Thomas, 1980), и частыми являются спорадические случаи. Имеется заметное преобладание мужчин, особенно при мягких формах, что предполагает возможность Х-сцепленного наследования в некоторых семьях. Болезнь трудно отличить от невральных перонеальных атрофий, при которых признаки сенсорных нарушений вначале могут отсутствовать.
Поэтому критерии диагноза должны включать:
1) дистальную мышечную атрофию, обычно преобладающую в нижних конечностях;
2) отсутствие сенсорных аномалий;
3) соответствующую ЭМГ и патологические признаки поражения клеток передних рогов; и
4) период, по крайней мере, 18 месяцев без сенсорной симптоматики (Harding и Thomas, 1980).
Pes cavus, часто тяжелой степени, относится к частым признакам. Ахиллов рефлекс и сухожильные рефлексы верхних конечностей часто сохраняются в отличие от проявлений при болезни Шарко-Мари-Тута. Возраст начала варьирует от грудного до середины жизни и может отличаться внутри одной семьи (Boylan et al., 1995). Описаны случаи с картированием на участке хромосоме 12q24 (De Angelis et al., 2002). Обычно течение медленное, но прогрессирующее, пациентам к 25-30 годам требуются вспомогательные средства для передвижения, хотя во многих случаях инвалидизация во взрослом возрасте умеренная. При редких формах верхние конечности поражаются первыми и преимущественно с медленным течением (Timmerman et al., 1996; Hedge et al., 2001).
Эти формы предположительно наследуются доминантным путем или же проявляются спорадически. Сообщается о связи с зрительно-слуховой атрофией (Chalmers и Mitchell, 1987). Показано, что ранее описанная ассоциация дистальной СМА с параличом голосовой складки (Boltshauser et al., 1989) была вызвана скорее невропатией, а не заболеванием клеток передних рогов. Bertini et al. (1989) описали связь дистальной СМА с диафрагмальным параличом.
г) Лопаточно-перонеальная спинальная мышечная атрофия (СМА). Лопаточно-малоберцовый синдром, характеризующийся истощением и слабостью в плечевом поясе и передней группе мышц ноги, может быть вызван миопатическим, невральным поражением или вовлечением клеток переднего рога. Последний тип является наиболее распространенным (Mercelis et al., 1980). Он обычно наследуется по аутосомно-доминантному типу, но могут встречаться рецессивное наследование и Х-сцепленная передача (Thomas et al., 1972). Связь с хромосомным локусом 12q24—31 была найдена в одной семье (Isozumi et al., 1996). Некоторые исследователи (например, Dubowitz, 1995) выразили сомнения относительно реальности СМА как причины лопаточно-перонеального синдрома.
Лице-лопаточно-перонеальная спинальная мышечная атрофия (СМА) является редким типом, имитирующим более частую лице-плече-лопаточную мышечную дистрофию. Наследование идет аутосомно-доминантным путем. Начало заключается в лицевой диплегии с прогрессирующим нисходящим распространением слабости (Furukawa и Toyokura, 1976). У одного ребенка дополнительно присутствовали атрофия зрительного нерва и сенсорные и вегетативные нарушения (Schmitt et al., 1994).
д) Доброкачественная семейная спинальная мышечная атрофия (СМА) с гипертрофией икроножных мышц. Гипертрофия икроножных мышц может быть сравнительно распространена при хронических СМА (Bertorini и Igarashi, 1985). Bouwsma и Van Wijngaarten (1980) выявили ее у 29 из 100 пациентов. Все были мужчинами, что наводит на мысль о возможной роли Х-сцепленного фактора. D’Alessandro et al. (1982) описали доминантную форму у отца и сына.
е) Нетрадиционные формы (паттерны) спинальной мышечной атрофии (СМА):
1. Мономелическая или ассиметричная СМА. Первые сообщения о мономелической или явно асимметричной форме прогрессирующей СМА получены из Японии, где это заболевание, вероятно, распространено (Hirayama et al., 1987), а впоследствии также из других стран (Tandan et al. 1990; Liu и Specht 1993; Restuccia et al., 2003). Болезнь преимущественно поражает мужчин, и клинические симптомы заболевания включают ювенильное начало, между 15 и 25 годами, незаметно начинающуюся прогрессирующую мышечную атрофию, не отягощенную инфекцией или травмой. Атрофия не вовлекает т. brachioradialis и связана со слабостью, усиливаемой холодом (холодовой парез) и с легким нерегулярным тремором.
Могут быть поражены обе стороны, почти всегда асимметрично. Денервационные изменения выявляются на ЭМГ и в биоптате мышцы, и часто являются двусторонними (Misra et al., 2005), даже если клиническое поражение кажется односторонним. Сенсорная патология отсутствует, скорость нервной проводимости в норме. Заболевание является обычно спорадическим, но известны несколько семейных случаев (Tandan et al., 1990). Патоморфологические изменения (Hirayama et al., 1987) включают сморщивание и некроз больших и малых мотонейронов. Несколько недавних исследований указывают на вероятность цервикальной миелопатии, связанной с венозной дилатацией при травматическом сгибании шеи (Hirayama и Tokumaru, 2000; Restuccia et al., 2003; Baba et al., 2004).
2. Сложные формы СМА. Ювенильная болезнь мотонейрона распространена в южной Индии. Наиболее точно определенный вариант известен как мадрасский тип. Заболевание обычно спорадическое; преобладает у мужчин и начинается в течение второго десятилетия жизни. Первоначально происходит поражение преимущественно верхних конечностей, хотя в итоге вовлекаются и нижние. Нейросенсорная тугоухость присутствует у трети пациентов, а бульбарное поражение обнаруживается приблизительно в 40% случаев (Gourie-Devi и Suresh, 1988). Болезнь является медленно прогрессирующей и весьма часто приостанавливается. Был описан вариант с атрофией зрительного нерва и поражением мозжечка (Gourie-Devi и Nalini, 2003).