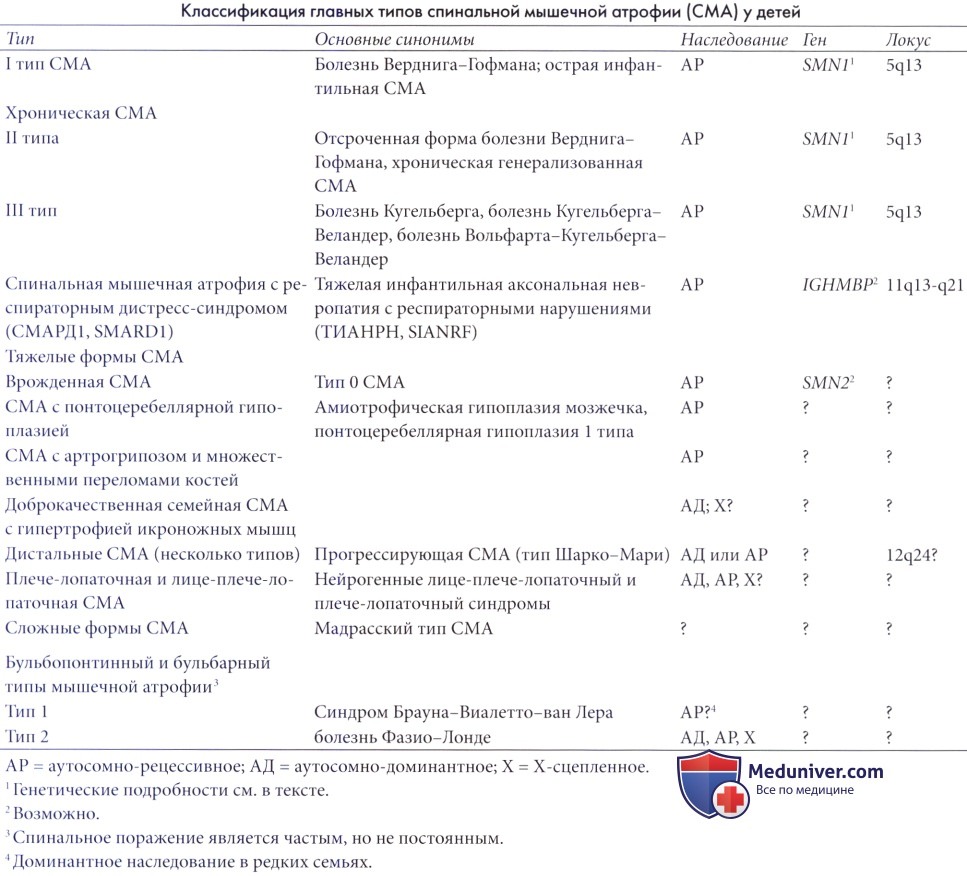
Хроническая спинальная мышечная атрофия III типа (СМА третьего типа, болезнь Вольфарта-Кугельберга-Веландер)
Хроническая спинальная мышечная атрофия III типа (СМА третьего типа, болезнь Вольфарта-Кугельберга-Веландер, псевдомиопатическая СМА, болезнь Кугельберга, болезнь Кугельберга-Веландер) также передается по аутосомно-рецес-сивному типу, хотя нередки и спорадические случаи. Она также в большинстве случаев связана с делецией в гене SMN1. Заболевание может начаться в любой момент грудного возраста или раннего детства и является чрезвычайно коварным.
Часто различают два подтипа: пациенты с подтипом IIIa начинают ходить в возрасте до трех лет, а пациенты с подтипом IIIb — после этого возраста, и данное различие может иметь отношение к прогнозу (Rudnik-Schoneborn et al., 2001).
Родители могут сначала заметить небольшой объем мышц бедра ребенка или отметить трудности при вставании с пола. Слабость и амиотрофия явно преобладают в проксимальном сегменте нижних конечностей и могут избирательно вовлечь m. quadriceps. При исследовании выявляются положительный симптом Говерса, истощение дистальных мышц и отсутствие коленных рефлексов с сохранением ахилловых рефлексов и рефлексов с верхних конечностей. Налицо клиническое сходство с мышечными дистрофиями с «утиной» походкой. У некоторых пациентов может присутствовать гипертрофия икроножных мышц, завершая сходство с дистрофией Дюшенна (Bouwsma и Van Wijngaarden, 1980).
Болезнь прогрессирует очень медленно в направлении дистальных отделов нижних конечностей и/или проксимальных отделов верхних конечностей. Часто встречается полая стопа. Многие пациенты демонстрируют грубый тремор кистей (Dawood и Moosa, 1983), который, вероятно, связан с несовершенной синхронизацией уменьшенного числа больших моторных единиц в результате коллатеральной реиннервации мышечных волокон оставшимися нейронами. ЭКГ показывает дрожание изолинии, по-видимому, отражающее тот же самый мышечный тремор.
Наиболее пострадавшие люди могут продолжить вести нормальную жизнь во взрослом возрасте (Zerres et al., 1995, 1997), но некоторые случаи протекают быстрее; ортопедические осложнения могут произвести к тяжелым функциональным ограничениям, которых желательно избегать в ходе адекватной физиотерапии и/или ортопедического лечения. Правильное консультирование с настоятельной рекомендацией приобретения навыков, позволяющих самостоятельное проживать длительное время, является крайне важным. В редких случаях прослеживается связь с врожденной (Oka et al., 1995) или прогрессирующей (Alberfeld и Namba, 1969) офтальмоплегией.
Отличие СМА III типа от мышечной дистрофии бывает затруднено, поскольку клиническая картина нередко наводит на мысли о последней, а уровень креатинкиназы может быть умеренно повышен. Биопсию мышцы иногда трудно интерпретировать из-за наличия таких «миопатических» изменений, как расщепленные или некротизированные волокна, центральные ядра и некоторый избыток соединительной и жировой ткани. ЭМГ — лучшая диагностическая методика, достаточная для диагностики, по мнению автора этих строк.
Помимо миогенных заболеваний, дифференциальный диагноз также включает редкие заболевания, такие как ганглиозидоз «взрослого типа», который может проявляться изолированным поражением клеток передних рогов (Navon et al., 1995), и исключительные случаи интрамедуллярных опухолей, проявляющихся как изолированная СМА (Aysun et al., 1993).
Лечение СМА III типа основано на физиотерапии и профилактике контрактур и ретракций. Для обоих типов проведено несколько испытаний препаратов, способных вызвать изменения в чтении ДНК. Они включают габапентин (Miller et al., 2001; Merlini et al., 2003), рилузол (Russman et al., 2003), фенилбутират (Mercuri et al., 2004a). Несмотря на некоторые положительные результаты, ни один из них не доведен до стадии практического применения. Генная терапия находится в стадии изучения (DiDonato et al., 2003).
Молекулярно-генетический диагноз рецессивных проксимальных СМА. Диагностика трех классических типов СМА методами молекулярной генетики возможна главным образом за счет демонстрации гомозиготной делеции 7 экзона в гене SMN1 (Melki et al., 1994). Однако делеция отсутствует в меняющейся, но немногочисленной группе пациентов с СМА; редкие люди с гомозиготной делецией могут иметь нормальный фенотип; и отношение генов SMN к СМА еще не полностью определено.
Приблизительно 10% случаев СМА (например, СМАРД1, СМА с оливопонтоцеребеллярной атрофией), не связаны с делецией или связаны с 5 хромосомой (Novelli et al., 1995; Rudnick-Schoneborn et al., 1995). Напротив, ряд случаев с наличием одного или нескольких принятых критериев исключения (Munsat и Davies, 1992) может демонстрировать типичную делению, включая случаи с дисфункцией центральной нервной системы, артрогрипозом или сочетанными пороками развития (Rudnik-Schoneborn et al., 1996).
Анализ ДНК обеспечивает возможность достоверного пренатального диагноза большинства типов классической СМА (Cobben et al., 1993; Wirth et al., 1995). Можно провести тест ДНК с использованием ПЦР (Van der Steege et al., 1995). Суть теста заключается в демонстрации гомозиготной делеции 7 экзона гена SMN. Он относительно легко выполним, но может быть негативным приблизительно у 2% пораженных детей, которые являются носителями отличающегося дефекта ДНК (Raymond, 1997).