Атипичные и пограничные формы болезни Верднига-Гофмана (СМА первого типа)
а) Спинальная мышечная атрофия с респираторным дистресс-синдромом 1 типа (здесь и далее — СМАРД1, англ. SMARD1). СМАРД1 спинальная мышечная атрофия с параличом диафрагмы с ранним началом была первоначально расценена как атипичная форма СМА (McWilliam et al., 1985), несмотря на обычную сохранность диафрагмальной функции при СМА. Фактически, это состояние возникает из-за другого генетического расстройства с мутациями в гене иммуноглобулина μ связывающего белка 2 (IGHMBP2) на участке хромосомы 11q13-q21.
Были описаны несколько различных мутаций, показывающих аллельную гетерогенность (Мауstadt et al., 2004). Болезнь наследуется аутосомно-рецессивным путем и представляет приблизительно 1% случаев СМА с ранним началом. Она клинически гетерогенна, с вариабельным течением. В большинстве случаев (Viollet et al., 2002; Grohmann et al., 2003; Pitt et al., 2003; Rudnik-Schoneborn et al., 2004) клиническое начало в течение первых месяцев жизни проявляется генерализованной гипотонией, слабым криком, стидором и деформацией стоп.
Задержка внутриутробного развития указывает на патологическое пренатальное развитие у трех четвертей младенцев, а более трети детей рождаются преждевременно. Диафрагмальный паралич обычно очевиден к возрасту от шести месяцев до одного года, часто с необходимостью постоянной вентиляции. Быстро проявляются прогрессивная дистальная мышечная атрофия и суставные контрактуры. Подозрения вызывают свисающие кисти и жировые прослойки подушечек пальцев. Слабость мышц лица не типична. Часто присутствуют признаки периферического поражения сенсорных и вегетативных нервных волокон, и это состояние также рассматривается некоторыми авторами как тип периферической невропатии (Pittet al., 2003; Duval-Beaupere et al., 1994, Diers et al., 2005).
Возможны нарушения опорожнения кишечника и мочевого пузыря, и у некоторых детей могут быть найдены признаки вегетативной дисфункции в виде аритмии сердца, артериальной гипертензии и чрезмерного потоотделения. Снижение скорости проведения и ЭМГ-признаки денервации присутствуют у большинства детей. Болезнь развивается медленно, но респираторный дистресс представляет опасность, и в некоторых случаях может развиться слабость бульбарной группы мышц (Rudnik-Schoneborn et al., 2004). Возможен пренатальный диагноз (Grohmann et al. 2003, Sangiuolo et al., 2004).
Тем не менее, возраст начала и степень тяжести могут быть различными даже в пределах одной и той же семьи, включая некоторых пациентов, имеющих нормальную мышечную силу вплоть до взрослого возраста и нормальную функцию диафрагмы во взрослый период жизни (Viollet et al., 2002).
б) Амиотрофическая понтоцеребеллярная гипоплазия. Тяжелая форма врожденной СМА наблюдалась в нескольких семьях в сочетании с понтоцеребеллярной гипоплазией. У таких пациентов также имеют место тяжелая задержка в развитии, поражение клеток передних рогов, признаки периферического поражения нерва с заметно сниженной моторной скоростью проведения (Goutieres et al., 1977; Kamoshita et al., 1990). Это состояние, сочетающее признаки как спиноцеребеллярной дегенерации, так и порока развития червя и передней доли мозжечка с тяжелой гипоплазией, поражающей ствол мозга, называется также инфантильной нейронной дегенерацией (Steinman et al., 1980), болезнью Нормана (Kamoshita et al., 1990) и понтоцеребеллярной гипоплазией 1 типа (Barth, 1993).
Болезнь проявляется в разгибательных контрактурах и часто быстро приводит к летальному исходу. Однако некоторые пациенты могут демонстрировать более умеренные симптомы и выживать до нескольких лет (Muntoni et al, 1999, Rudnik-Schonebornet al., 2003a). Нарушение не сцеплено с 5 хромосомой.
в) Спинальная мышечная атрофия (СМА) с артрогрипозом и множественными переломами костей. Заболевание нередко с быстрым смертельным исходом (Van Toorn et al., 2002, Felderhoff-Mueser et al., 2002), как и два предыдущих не связанное с делецией в гене SMN во всех случаях (Rudnik-Schoneborn et al., 1996), хотя Burglen et al. (1996) продемонстрировали делецию 7 экзона у шести из 12 младенцев, страдающих СМА с артрогрипозом, a Garcia-Cabezas et al. (2004) сообщили о ребенке с переломами и делецией гена SMN1.
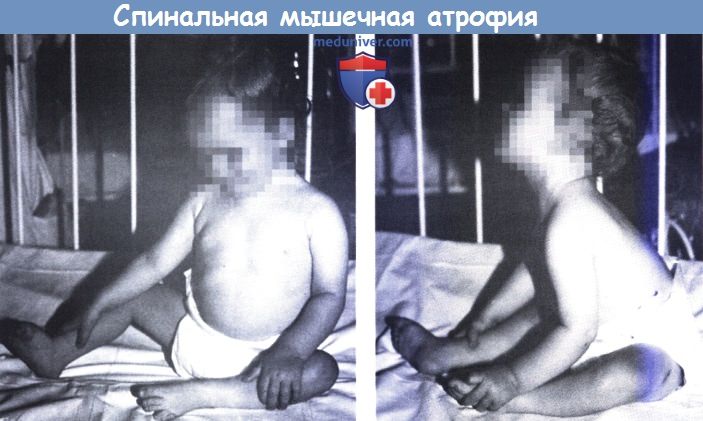
Спинальная мышечная атрофия с преобладанием на уровне шеи (14-месячная девочка с полным параличом как передних,
так и задних шейных мышц с сохранением нормальной силы в нижних конечностях, но с некоторой слабостью и гипотрофией в верхних конечностях).
Голова наклонена вперед, больная не способна удерживать ее прямо (слева).
После запрокидывания назад голову не удается вернуть в переднее положение (справа).
г) Спинальная мышечная атрофия (СМА), первоначально ограниченная или явно преобладающая в цервикальных мышцах. Некоторые случаи прогрессирующей СМА, поражающей первоначально и преимущественно мышцы шеи, могут отличаться от классической болезни Верднига-Гофмана (Goutieres et al., 1991). Больные дети способны ходить. Вторичное распространение идет по нисходящей с параличом межреберных мышц, который, в конечном счете, приводит к нарушению дыхания и смерти. Исследование ДНК в указанных случаях было недоступно.
д) Прогрессирующая спинальная мышечная атрофия (СМА) с офтальмоплегией и пирамидными симптомами. Данное сочетание наблюдалось у двух сибсов (Нашапо et al., 1994). Было показано, что подобные случаи с поражением моторных и сенсорных нервов и быстрым летальным исходом, имели связь с большой делецией, затрагивающей ген SMN и несколько фланкирующих маркеров (Korinthenberg et al, 1997).
Такие атипичные формы достоверно представлены в нескольких семьях, что показывает, что СМА — негомогенная группа.
е) Форма с острым началом. Описаны формы с острым началом после интеркуррентных инфекций (Robb et al., 1991).