Нервная система при нарушениях в дыхательной цепи (цикле Кребса)
В связи со сложностью генетических и биохимических характеристик митохондрий клинические проявления данных заболеваний чрезвычайно разнообразны. Проявления варьируют от единичных поражений тканей или органов (нейропатия зрительного нерва при наследственной болезни Лебера) до более распространенных поражений, включающих миопатии, энцефалопатии, кардиомиопатии или сложные мультисистемные синдромы, проявляющиеся в период от новорожденности до взрослого возраста.
Определенные сочетания длительное время классифицировались как отдельные заболевания, такие как синдромы Кирнса-Сайера, MELAS, MERFF, NARP, Ли и болезнь Альперса. Тем не менее, не всех пациентов с нарушениями дыхательной цепи можно так легко отнести к какой-то группе, зарегистрированы перекрестные проявления различных групп заболеваний. Следовательно, нарушения дыхательной цепи следует предполагать у пациентов с необъяснимыми прогрессирующими нервно-мышечными и не нервно-мышечными симптомами (DiMauro и Schon, 2003; Zeviani и Di Donato, 2004; Robinson, 2006).
а) Миопатические формы. Миопатические формы характеризуются прогрессирующей слабостью конечностей с различной степенью непереносимости нагрузок, мышечной болью, одышкой, утомляемостью и тошнотой. Симптомы могут появляться в течение первых двух лет жизни в виде гипотонии и отсрочки появления двигательных навыков или в более позднем детском возрасте, подростковом или взрослом возрасте. Смерть может наступать в результате ранней сердечно-легочной недостаточности или же сохраняется стабильное состояние в течение нескольких лет. Содержание лактата в крови может быть в пределах нормы или несколько повышаться.
Кратковременные аэробные нагрузки могут провоцировать развитие тяжелого лактатацидоза. За исключением очень маленьких, обычно характерны морфологические и гистохимические аномалии, выявляемые при биопсии мышц. Данные изолированные миопатические формы сочетаются с точечными мутациями мтДНК в генах, кодирующих субъединицы комплексов I, III (цитохром b) или IV. Дефект, вызванный мутациями гена цитохрома b, вероятно, является наиболее характерным примером, так как преимущественно проявляется изолированной прогрессирующей миопатией и непереносимостью нагрузок. Редкие случаи заболевания в младенческом или подростковом возрасте проявляются преимущественным поражением мышц, к которому в конечном счете присоединяется поражение других органов, вызванное мутациями генов тРНК или митохондриальной деплецией (Alberio et al„ 2007).
Первичный дефицит кофермента Q10 (аутосомно-рецессивное заболевание с широким спектром клинических проявлений) проявляется чистой ювенильной миопатической формой с непереносимостью нагрузок и миоглобинурией. Другие проявления включают энцефаломиелопатию с поражением головного мозга и рецидивирующей миоглобинурией, тяжелое младенческое мультисистемное поражение, мозжечковую атаксию, синдром Ли с атаксией и глухотой. Такие пациенты с разорванными красными волокнами и отсутствием кофермента Q10 в тканях поддаются лечению коферментом Q10 в высоких дозах. При младенческой мультисистемной форме выявлена мутация гена COQ2. Данный ген кодирует первый фермент, необходимый для биосинтеза кофермента Q10.
Миопатические проявления дефицита цитохром С оксидазы (комплекса IV) включают три основных формы: смертельная младенческая миопатия, доброкачественная обратимая младенческая миопатия и миопатия с поздним началом (миопатия взрослых). Смертельная и доброкачественная формы заболевания проявляются у новорожденных тяжелой генерализованной слабостью, нарушением дыхания и лактатацидозом. Пациенты со смертельной формой заболевания обычно умирают от дыхательной недостаточности в течение нескольких месяцев. Тем не менее, имеются сведения о пациентах, доживших до семи лет, имеющих инвалидность в связи с непереносимостью физических нагрузок, но нормальное умственное развитие и способность к обучению.
С другой стороны, у пациентов с доброкачественной формой заболевания отмечается спонтанное улучшение, несмотря на исходную выраженную слабость, и ранний детский возраст обычно протекает нормально. Некоторые тяжелые формы, имитирующие спинальную мышечную атрофию, описаны при мутациях ядерного гена SC02, митохондриального гена СОХ1 и деплеции мтДНК в связи с мутациями ядерного гена, кодирующего тимидинкиназу 2 (ТК2).
б) Мультисистемные поражения. При данных заболеваниях поражается преимущественно ЦНС, а проявления возникают в периоде новорожденности или позднее в детском, подростковом или даже взрослом возрасте.
1. Смертельная младенческая форма. Данная форма заболевания проявляется лактатацидозом и гипотонией новорожденных, припадками и нарушениями дыхания; следствием заболевания является смерть, наступающая через несколько месяцев. Как и при дефиците комплекса IV может отмечаться почечная, сердечная и печеночная недостаточность. Выявляется тяжелая гиперлактатемия, в то время как результаты морфологических исследований мышц могут не отличаться от нормы или свидетельствовать о неспецифическом накоплении липидов и гликогена. На КТ и МРТ можно обнаружить корковую или подкорковую атрофию и дисмиелинизацию. Данный синдром встречается часто, но не является единственным дефектом, а сочетается с дефицитом комплекса I и комплекса IV.
2. Прогрессирующая энцефалопатия/энцефаломиелопатия. Тяжелая энцефалопатия может начинаться в позднем младенческом или в детском возрасте после периода практически нормального развития. Такие «неопределенные» типы заболевания могут клинически проявляться варьирующими компонентами следующих состояний: задержкой развития, гипотонией, слабостью, атаксией, пирамидными знаками, припадками, миоклонией, ретинопатией, птозом, прогрессирующей внешней офтальмоплегией, нейросенсорной тугоухостью, легким дисморфизмом и задержкой роста. Уровень лактата в плазме умеренно повышен. В таких случаях высокий уровень лактата в спинномозговой жидкости и лактатурия имеют высокое диагностическое значение.
У некоторых пациентов отмечаются приступы тошноты после интеркуррентных заболеваний, во время которых отмечается высокий уровень лактата в плазме и можно определить соотношение окисления и восстановления. Наличие разорванных красных волокон часто сочетается с накоплением липидов-гликогена. Результаты КТ и МРТ могут не отличаться от нормы или свидетельствовать об атрофии головного мозга и/или мозжечка и двустороннем снижении плотности базальных ганглиев или белого вещества. Могут выявляться отклонения по результатам ЭМГ, изменения нервной проводимости и вызванных зрительных потенциалов или потенциалов ствола мозга. Описаны случаи дефектов всех комплексов дыхательной цепи вследствие различных точечных мутаций в мтДНК или яДНК.
Специфические сочетанные неврологические признаки, такие как полимикрогирия или сочетанные висцеральные проявления или гепатопатия могут способствовать постановке верного диагноза.
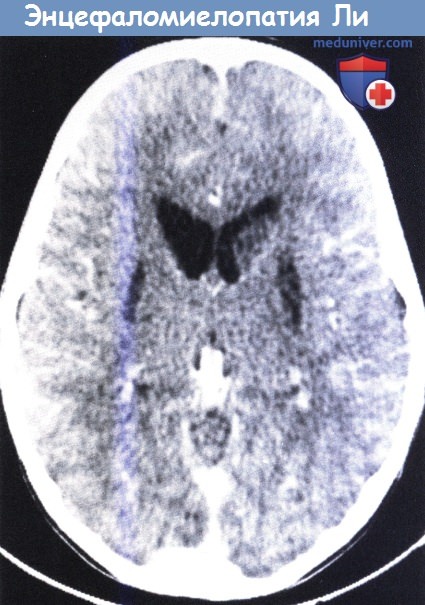
3. Подострая некротизирующая энцефалопатия: синдром Ли. Синдром Ли является наследственным прогрессирующим метаболическим заболеванием младенческого и детского возраста. Заболевание является причиной невропатологических проявлений в виде очаговых, двусторонних и симметричных некротических повреждений, сочетающихся с демиелинизацией, пролиферацией сосудов и глиозом ствола мозга, промежуточного мозга, базальных ганглиев, мозжечка и, в отдельных случаях, белого вещества (Farina et al, 2002). Явная связь между синдромом Ли и «неопределенными» энцефаломиелопатиями не прослеживается.
Нейропатологические изменения чаще всего затрагивают ствол мозга с двух сторон и распределяются приблизительно симметрично. Повреждения резко очерчены и пересекают серое и белое вещество, преобладая в первом. Патологический комплекс при синдроме Ли включает заметное губчатое поражение, преимущественно затрагивающее нейропиль, в то время как нейроны остаются относительно неповрежденными. Повреждения белого вещества включают утрату миелина и в итоге разрушение аксонов. Необходимым проявлением является интенсивная капиллярная пролиферация с отеком эндотелия.
Повреждения обычно имеют ишемический характер, но не соответствуют зоне кровоснабжения (Cavanagh и Harding, 1994). Для данного типа повреждений характерно поражение покрышки среднего мозга и моста, околоводопроводного серого вещества, черной субстанции и задних бугорков, дна четвертого желудочка и зубчатых ядер мозжечка. Часто встречаются повреждения базальных ганглиев, в особенности скорлупы и хвостатого ядра. Повреждения коры носят выраженный характер только в редких случаях. Экстенсивная лейкоэнцефалопатия выявляется редко (Bourgeois et al., 1992; Zafeiriou et al, 1995).
Мамиллярные тельца поражаются редко, что важно для дифференцировки заболевания от болезни Вернике, при которой отмечаются сходные изменения, но всегда поражаются данные структуры.
Клинические проявления чрезвычайно вариабельны, а диагностика заболевания сложна, так как в большинстве случаев отсутствуют биохимические маркеры. Некоторые частые проявления, такие как ухудшение с медленным восстановлением после инфекций, непереносимость нагрузок, замедленный рост и резкие изменения частоты дыхания или сердечных сокращений позволяют предположить наличие митохондриального заболевания.
Синдром Ли.
Типичная гистологическая картина: видны четко очерченные области выраженной губчатой структуры с интенсивной капиллярной пролиферацией.
При младенческой форме признаки заболевания обычно появляются в течение первых лет жизни и включают гипотонию, пирамидные знаки, плохое прибавление в весе, регресс психомоторного развития и дисфункцию базальных ганглиев. Поражение базальных ганглиев приводит к атаксии, аномальным движениям глаз, таким как птоз или офтальмоплегия, дистонии или ригидности и нарушениям глотания. Нарушения движений, включающие хореоатетоз, дистонию и в некоторых случаях миоклонус, не так уж редки. Могут возникать эпизоды ступора или апноэ, не связанного со ступором. Нередко отмечается поражение периферической нервной системы (Jacobs et al., 1990).
В редких случаях такое поражение может стать основным проявлением, имитируя синдром Гийена-Барре. У некоторых пациентов отмечаются припадки и могут встречаться младенческие судороги. Заболевание часто стремительно прогрессирует с периодами ремиссий и обострений. Обострения порой провоцируются инфекциями или голоданием. Зарегистрированы случаи острого фульминантного течения, затяжного течения с удлиненными периодами стабилизации и случаи очень медленного прогрессирования.
Формы с поздним началом встречаются реже, но могут выявляться в течение детского и подросткового возраста, а также во взрослом возрасте. Преобладающими проявлениями могут быть экстрапирамидные симптомы, такие как дистония или аномальные движения, гипокинезия и ригидность. В некоторых случаях умеренная задержка психомоторного развития с легкими неврологическими аномалиями может сохраняться неизменной в течение нескольких лет до момента развития стремительной деградации, которая в некоторых случаях отмечается только во взрослом возрасте.
Несмотря на то, что исходные наблюдения предполагают аутосомно-рецессивное наследование, зарегистрированы случаи аутосомно-доминантного, сцепленного с Х-хромосомой наследования и наследования по материнской линии, что свидетельствует о наличии измененных генов как в ядерном, так и в митохондриальном геноме. Такая генетическая гетерогенность была подтверждена выявлением функциональных или молекулярных дефектов нескольких ферментов, участвующих в выработке энергии, включая пируватдегидрогеназу, пируват карбоксилазу и комплексы дыхательной цепи. Как уже указывалось для неидентифицированных энцефаломиелопатий, возможно сочетание некоторых специфических клинических, биохимических и генетических изменений, определяющих окончательный диагноз.
В младенческом возрасте дефекты каждого комплекса дыхательной цепи могут проявляться в виде синдрома Ли. Такая клиническая картина обычно является проявлением изолированного выраженного дефицита ЦОГ. В сочетании с периферической нейропатией это вызвано главным образом мутациями ядерного SURF1 гена. Синдром Ли также является обычным проявлением дефицита комплекса I, вторичного по отношению к мутациям субъединицы, кодируемой ядерным или митохондриальным геном. В сочетании с гипертрофической кардиомиопатией данный синдром может свидетельствовать о дефекте субъединицы, кодируемой ядерным геном (NDFS2, NDFS4, NDFS7 и NDFS8). Дефект комплекса II, вызванный мутациями гена SDFIA, является причиной синдрома Ли с характерным поражением белого вещества и выявляемым на МР спектроскопии пиком сукцината (Brockmann et al., 2005).
Мутация гена BCS1L, объединяющего гена комплекса III, проявляется в раннем младенческом возрасте в виде специфического сочетания синдрома Ли, печеночной недостаточности и тубулопатиии, в то время как делеция VII субъединицы (UQCRB) вызывает лактатацидоз с гипогликемией. Мутации митохондриального гена АТР6, кодирующего комплекс V, при высокой гетероплазмии являются причиной наследования синдрома Ли по материнской линии, а при низком титре проявляются нейрогенной слабостью, атаксией и пигментным ретинитом (синдром NARP). Кроме того, раннее начало синдрома Ли может сочетаться с синдромами митохондриальной деплеции, такими как SUCLA2 и POLG и сочетается в таких случаях с гепатопатией.
Типичные изменения, выявляемые на КТ и МРТ, необходимы для диагностики синдрома Ли в условиях in vivo, так как они могут быть более характерными проявлениями, чем сочетание определенных клинических проявлений и лактатацидоза. Изменения заметны при нейровизуализации и имеют вид гиперинтенсивных участков при протонной плотности и в Т2-режиме, в то время как в Т1-режиме они обычно менее заметны и имеют пониженную плотность. Изменения обычно обнаруживаются в стволе мозга, где они поражают различные структуры продолговатого мозга, моста и среднего мозга. Часто отмечается поражение мозжечка, сосредоточенное в основном в зубчатом ядре с распространением на окружающее белое вещество.
Третьей областью повреждения являются базальные ганглии, таламус и субталамическое ядро. Дополнительными проявлениями могут быть обширные повреждения глубокого белого вещества головного мозга, и прогрессирующая церебральная атрофия или инфаркты. Явная корреляция между специфическим ферментным дефектом и рентгенологическими изменениями не прослеживается. Данное обстоятельство частично объясняется изменениями повреждений ЦНС и тем, что дефект какой-либо структуры может быть вызван различными молекулярными повреждениями с различными последствиями. Тем не менее, специфическое сочетание тяжелого поражения ствола мозга и субталамических структур описано при синдроме Ли с дефицитом ЦОГ, вызванным мутациями гена SURF1 (Valanne et al., 1998; Arii и Tanabe 2000; Farina et al., 2002).
Некротизирующая энцефаломиелопатия Ли.
Кавитация обоих лентикулярных ядер. Пониженная плотность левого хвостатого ядра.
Имеется заметная атрофия головки правого хвостатого ядра с расширением соответствующего переднего рога.
4. Синдром Кирнса-Сейра. Синдром Кирнса-Сейра обычно представляет собой мультисистемное спорадическое заболевание, характеризующееся неизменной триадой, включающей начало в возрасте до 20 лет, прогрессирующую внешнюю офтальмоплегию и пигментную дегенерацию сетчатки, а также минимум одно из следующих проявлений: предсердно-желудочковую блокаду, повышение уровня белка в спинномозговой жидкости и дисфункцию мозжечка (Zeviani и Di Donato, 2004). Тем не менее, во многих случаях отмечаются и другие клинические аномалии, описано два постоянных патологических проявления: разорванные красные волокна и губчатая дегенерация головного мозга. Описанные признаки и симптомы могут прогрессировать в различной последовательности.
У детей с синдромом Кирнса-Сейра обычно отмечается нормальное раннее развитие. В детском возрасте иногда встречаются эпизоды асептического менингита. Птоз и прогрессирующая внешняя офтальмоплегия обычно являются первыми неврологическими проявлениями заболевания, часто сочетающимися с дегенеративной пигментной ретинопатией, а иногда с атрофией зрительного нерва. Наиболее частым неврологическим проявлением является мозжечковый синдром, который может приобретать выраженный характер. Возможна задержка или регресс умственного развития. Припадки не развиваются, за исключением случаев сопутствующей гипокальциемии с гипопаратиреозом. Слабовыраженные пирамидные знаки, миопатия или периферическая нейропатия не отчетливы.
Предсердно-желудочковая блокада является поздним признаком, который может стать причиной синкопальных состояний или внезапной смерти, несмотря на установку водителя ритма. В редких случаях развивается застойная сердечная недостаточность и суправентрикулярная тахикардия. Сочетанные эндокринные нарушения могут включать низкий рост с дефицитом гормона роста, латентный диабет и гипопаратиреоз. Нарушение функции почек часто включает дефект проксимальных трубочек, дистальную тубулопатию, гломерулопатию и почечную недостаточность. Типичными изменениями лабораторных показателей являются легкий лактатацидоз и высокий уровень белка в спинномозговой жидкости.
При нейровизуализации выявляются повреждения серого и белого вещества, чаще всего расположенные в покрышке ствола мозга, белом веществе головного мозга и мозжечка и базальных ганглиях. Часто отмечается кальцификация базальных ганглиев или глубоких слоев белого вещества. Поражение подкорковых U-волокон с относительным разрежением перивентрикулярного белого вещества типично для синдрома Кирнса-Сейра. Тем не менее, данные классические повреждения развиваются прогрессивно со временем, и МРТ на ранних стадиях заболевания может не отличаться от нормы (Valanne et al., 1998). Затухание электроретинограммы и аномальные вызванные зрительные потенциалы могут предшествовать офтальмоскопическим признакам ретинопатии. ЭМГ и скорость проводимости нерва в некоторых случаях могут указывать на периферическую нейропатию и миопатические изменения.
Гистохимические исследования и электронная микроскопия скелетных мышц позволяют выявить разорванные красные волокна и повреждение митохондрий. Повреждение волокон I типа в отсутствие гистохимических признаков активности цитохром С оксидазы сочетается с нормальными волокнами. У 80% пациентов с синдромом Кирнса-Сейра имеется единичная делеция мтДНК. Тем не менее, встречается синдром Кирнса-Сейра без делеции и не у всех пациентов с делецией мтДНК развивается синдром Кирнса-Сейра. Чаще всего данное заболевание встречается спорадически, и только у немногих пациентов выявляется наследование по материнской линии.
Синдром Пирсона, который может возникать в том же возрасте, что и синдром Кирнса-Сейра, характеризуется врожденной сидеробластной анемией и, в более редких случаях, тяжелым нарушением экзокринной функции поджелудочной железы. Данный синдром представляет собой раннюю и часто смертельную форму синдрома Кирнса-Сейра со сходными единичными делециями/дупликациями мтДНК.
5. Прогрессирующая наружная офтальмоплегия. Прогрессирующая внешняя офтальмоплегия является клинически и генетически гетерогенным состоянием, сочетающимся с единичной или множественными делециями мтДНК и спорадическим, аутосомно-доминантным или аутосомно-рецессивным наследованием. В типичных случаях первые признаки возникают во взрослом возрасте, зарегистрировано небольшое количество случаев с началом в подростковом или раннем взрослом возрасте. Основными признаками являются офтальмоплегия, птоз и мышечная слабость с непереносимостью нагрузок. Другие прогрессирующие симптомы включают двигательную невропатию, нейросенсорную глухоту, катаракту и поражение мозжечка. Может развиться когнитивная деградация с психиатрическими проявлениями.
Аналогично синдрому Кирнса-Сейра единичные делеции/дупликации мтДНК являются причиной лактатацидоза, разорванных красных волокон и мозаичности дефекта цитохром С оксидазы по результатам гистохимических исследований мышц. Данные, связанные с синдромом Кирнса-Сейра, случаи чаще всего носят спорадический характер.
Аутосомно-доминантные формы прогрессирующей внешней офтальмоплегии в основном вызваны мутациями трех ядерных генов: POLG1, twinkle и ANT1. Мутации сопровождаются разорванными красными волокнами в скелетных мышцах и множественными делециями мтДНК. Типичными проявлениями аутосомно-доминантной прогрессирующей внешней офтальмоплегии являются мышечная слабость, сильнее всего поражающая внешние глазные мышцы. Другие варьирующие проявления включают атаксию, депрессию, гипогонадизм, тугоухость, периферическую нейропатию и катаракту. Другие случаи спорадической или рецессивной прогрессирующей внешней офтальмоплегии могут быть вызваны мутациями гена POLG (Zeviani и Di Donato, 2004; Sharer, 2005).
6. Миоклоническая эпилепсия с разорванными красными волокнами. Миоклоническая эпилепсия с разорванными красными волокнами представляет собой энцефаломиелопатию с наследованием по материнской линии, характеризуемую миоклонусом, мозжечковой атаксией и митохондриальной миопатией. Часто отмечаются припадки, тугоухость, деменция, периферическая нейропатия и множественные симметричные липомы (Zeviani и Di Donato 2004).
Обычно симптомы появляются в возрасте 5-13 лет и включают мозжечковую атаксию, тремор, миоклонические судороги, индуцируемые движениями или намеренными движениями. У многих пациентов развиваются генерализованные или тяжелые миоклонические припадки, а у некоторых формируется деменция. Может встречаться тугоухость, атрофия зрительного нерва и утрата проприоцептивной чувствительности. Прогрессирующая внешняя офтальмоплегия, дегенерация сетчатки или инсультоподобные эпизоды отсутствуют. Выраженность симптомов среди родственников по материнской линии широко варьирует от тяжелых до легких проявлений, иногда отмечаются только аномалии вызванных потенциалов и параметров электроретинограммы.
Возможно небольшое повышение лактата в спинномозговой жидкости и сыворотке. Могут встречаться эндокринные нарушения.
При записи электроретинограммы отмечается аномальная фоновая активность, различные сочетания с комплексами «пик-волна» и фотопароксизмальными реакциями. При оценке зрительных, слуховых и соматосенсорных вызванных потенциалов может выявляться аномальный период задержки, а по результатам ЭМГ отмечаются миопатические изменения. КТ и МРТ могут свидетельствовать об атрофии головного мозга и мозжечка. При патологоанатомическом исследовании выявляется дегенерация полушарий мозга, зубчатого ядра мозжечка, заднего столба спинного мозга и спиноцеребеллярного тракта.
Разорванные красные волокна с дефицитом ЦОГ и ультраструктурные аномалии митохондрий, выявляемые при биопсии мышц, являются постоянными проявлениями и могут являться единственным изменением у членов семьи с бессимптомным течением. Типичные мутации мтДНК отмечаются в генах A8344G на участке тPHKlys. Другие мутации тех же генов были зарегистрированы в сочетании с синдромом MERRF, MERRF/MELAS и другими комплексными фенотипами.
7. Митохондриальная миопатия, энцефалопатия, лактатацидоз и инсультоподобные эпизоды (синдром MELAS). Синдром MELAS обычно встречается у детей или молодых взрослых и развивается после нормального раннего развития. Симптомы включают рецидивирующую рвоту, мигренеподобные боли и рецидивирующие инсультоподобные эпизоды, приводящие к корковой слепоте, гемипарезу или гемианопсии. Припадкам часто предшествуют инсульты или приступы мигрени с аурой, припадки во всех случаях имеют парциальный моторный характер. Заметными проявлениями являются регресс интеллекта и поведенческие проблемы. Миопатия обычно протекает бессимптомно или выражается мышечной слабостью и легкой амиотрофией. Дополнительные проявления включают низкий рост, сахарный диабет, нейросенсорная тугоухость, легкую дегенерацию сетчатки и поражение сердца.
Уровень лактата в плазме и спинномозговой жидкости обычно высокий, уровень креатинкиназы повышался независимо. Содержание белка в спинномозговой жидкости не отличалось от нормы.
Параметры ЭЭГ обычно отличаются от нормы и характеризуются эпизодическими пиками и волнами или очаговыми пиковыми разрядами. При нейровизуализации выявляются очаги усиления сигнала в Т2-режиме, в целом без соответствия распределению сосудов, множество пораженных участков белого вещества, коры или ствола мозга, и области кальцификации или высокого сигнала в Т2-режиме в базальных ганглиях. Возможна сопутствующая атрофия мозга и мозжечка. Гистопатологические изменения включают очаговый некроз и пластинчатые некротические изменения коры, а также нейрональную дегенерацию и накопление минералов внутри базальных ганглиев. Патогенез инфарктоподобных изменений предположительно связан с митохондриальной микроангиопатией с дефицитом энергетического метаболизма в мелких артериолах и капиллярах мягкой мозговой оболочки (Valanne et al., 1998). В мышцах выявляются разорванные красные волокна, вкрапления жира и аномальные митохондрии.
Наиболее распространенной мутацией мтДНК является изменение гена A3243G в тPHKlyu(UUR)- Однако синдром MELAS связан со множеством других мутаций. С другой стороны, данная мутация гена A3243G была выявлена у пациентов с наследуемой по материнской линии прогрессирующей внешней офтальмоплегией, сахарным диабетом или тугоухостью и изолированной миопатией и кардиомиопатией.
8. Болезнь Альперса. Синдром Альперса-Хуттенлохера представляет собой аутосомно-рецессивный гепатоцеребральный синдром с началом в раннем возрасте, это дегенеративное заболевание с первичным поражением серого вещества головного мозга. Типичное течение синдрома Альперса-Хуттенлохера включает тяжелую задержку развития, некупируемые припадки, корковую слепоту, прогрессирующую печеночную недостаточность и острую печеночную недостаточность после приема вальпроевой кислоты; смерть наступает в детском возрасте. Отмечаются биохимические изменения, включающие дефицит пируваткарбоксилазы, пируватдегидрогеназы и компонентов дыхательной цепи.
Важно отметить, что синдром Альперса-Хуттенлохера является клиническим фенотипом, возникающим при синдроме митохондриальной деплеции, вызванной мутациями гена POLG1, приводящими к деплеции мтДНК и множественным дефектам дыхательной цепи во всех пораженных тканях (Gordon 2006; Horvath et al., 2006). Клинические проявления описаны в главе 9. Припадки и аномалии ЭЭГ значительно более выражены, чем при синдроме Ли, что является отражением экстенсивного поражения коркового серого вещества. Возможно сочетанное поражение различных внутренних органов.
9. Митохондриальная нейрогастроинтестинальная энцефалопатия (MNGIE). Синдром MNGIE представляет собой аутосомно-рецессивное заболевание, характеризуемое нарушением моторики желудочно-кишечного тракта, кахексией, птозом, офтальпопарезом, периферической нейропатией и изменениями белого вещества по результатам МРТ головного мозга.
Данное разрушительное заболевание связано с ядерными мутациями в гене тимидинфосфорилазы. Мутации индуцируют патологическое накопление тимидина и деоксиуридина и дисбаланс митохондриального депо, ухудшающий репликацию мтДНК, и/или приводящий к усилению репликации с деплециями, множественными делециями и точечными мутациями (Nishino et al., 2001).
Синдром MNGIE обычно манифестирует в позднем взрослом возрасте нарушением моторики желудочно-кишечного тракта в виде дисфагии, гастропареза и рецидивирующей рвоты, приводящей к кахексии. Данные признаки могут быть связаны с висцеральной миопатией, сильнее всего поражающей внешний слой собственно мышечной оболочки (Giordano et al., 2006). Неврологические изменения включают птоз и офтальмопарез с поздним началом и прогрессирующую аксональную и демиелинизирующую сенсомоторную полиневропатию. Когнитивные изменения обычно отсутствуют. Лактатацидоз обычно выражен слабо, а уровень белка в спинномозговой жидкости у большинства пациентов повышен. На МРТ в Т2 и FLAIR режимах выявляются распространенные сливающиеся гиперинтенсивные изменения сигнала, свидетельствующие о лейкоэнцефалопатии. Изменения локализуются преимущественно в околожелудочковых пространствах и могут поражать ножки мозга, ствол и мост.
в) Лечение. В настоящее время эффективные методы лечения нарушений дыхательной цепи отсутствуют. Лечение остается в основном поддерживающим, не оказывая влияния на течение заболевания.
В рамках симптоматической терапии не следует применять для лечения припадков вальпроаты, которые ингибируют дыхательную цепь.
Первичный дефицит кофермента Q10 можно лечить убидекареноном в высоких дозах, что приводит к заметному улучшению клинического течения. Тем не менее, данного лечения может быть недостаточно для предотвращения неврологической деградации (Aure et al„ 2004).
При синдроме MNGIE было предпринято несколько попыток восстановить нормальный уровень тимидина с помощью диализа, трансфузии тромбоцитов и даже трансплантации костного мозга.
Помимо описанных специфических методов используются различные «коктейли» витаминов и кофакторов, включая кофермент Q10, филлоквинон или менадион в сочетании с аскорбатом, сукцинатом, тиамином и рибофлавином. Применение дихлорацетата для лечения лактатацидоза не приводит к улучшению клинического исхода (Stacpoole et al., 2006). Поддерживающее применение креатина для обеспечения накопления креатинфосфата (энергетического вещества) оказывает различное воздействие (DiMauro et al., 2004а).