Нервная система при нарушениях митохондриального окисления жирных кислот
Врожденные нарушения митохондриального окисления жирных кислот представляют собой группу заболеваний, влияющих на энергетический метаболизм во время голодания и метаболического стресса. Вследствие этого типичные проявления включают острую метаболическую декомпенсацию, возникающую на фоне голодания, рецидивирующие гипогликемии, синдромы, подобные синдрому Рея, и необъяснимую внезапную младенческую смерть. Хроническое поражение тканей, зависимых от жирных кислот, может приводить к миопатии и кардиомиопатии. Известно около 20 различных дефектов (Wanders et al., 1999; Tein 2002; Vockley, 2002; Longo et al., 2006).
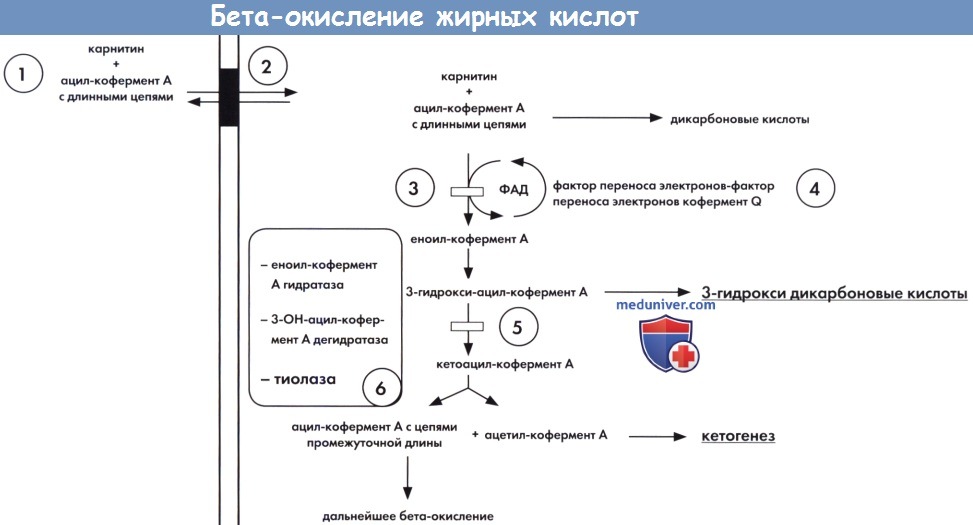
а) Биохимические изменения. Общие пути митохондриального окисления жирных кислот и различные известные дефекты ферментов представлены на рисунке ниже. Длинноцепочечные жирные кислоты, трансформированные в соответствующие эфиры кофермента А, транспортируются в митохондрии с помощью специфического карнитинового переносчика ацил-карнитина, который включают карнитин-пальмитоилтрансферазу I и II (КПТ-1 и КПТ-II) и карнитин-ацил-карнитин транслоказу (КАТ). Ацилкофермент А со средними и короткими цепями легко диффундирует в митохондрии. Первичный дефицит карнитина, КПТ-I, КПТ-II и транслоказы нарушают митохондриальный транспорт длинноцепочечных жирных кислот и следовательно, бета-окисление и кетогенез.
Дальнейшее бета-окисление затрагивает четыре последовательных этапа с участием ферментов, специфичных к длине цепи. На первом этапе необходима специфическая ацил-кофермент А дегидрогеназа, дегидрогеназы очень-длинно-, длинно-, умеренно-длинно- и короткоцепочечного ацил-кофермента A (VLCAD, LCAD, MCAD, SCAD), которые трансформируют жирные кислоты ацил-кофермента А в еноил-кофермент А. Все четыре дегидрогеназы высвобождают электроны, которые проходят через систему транспорта электронов дыхательной цепи. Специфическая система переноса электронов распределена между другими дегидрогеназами флавопротедов: глутарил-кофермент А и изовалерил-кофермент А дегидрогеназами.
Три оставшихся этапа бета-оксиления длинноцепочечных жирных кислот могут протекать под действием единственного трехфункционального белка (ТФБ) для субстратов с длинными цепями или под действием монофункциональных ферментов — для субстратов с короткими и умеренными цепями. Каждый четырехэтапный цикл бета-окисления приводит к высвобождению ацетил-кофермента А, который участвует в цикле Кребса в таких тканях, как сердце и мышцы. В печени в случае голодания ацетил-кофермент А превращается в кетоны (кетогенез), которые переносятся в периферические органы, такие как мозг, для окончательного окисления (кетолиза) и высвобождения энергии. Известные дефекты митохондриального бета-окисления приведены в таблице ниже. Дефицит множественной ацил-кофермент А дегидрогеназы (МАД), также называемой глутаровой ацидурией II типа, вызван дефектом системы передачи электронов. Несколько альтернативных путей приобретают значимость при нарушении митохондриального бета-окисления.
Омега-оксиление в микросомах приводит к образованию характерных дикарбоновых кислот, а интрамитохондриальная конъюгация ацил-кофермента А с глицином и карнитином является важном механизмом детоксикации.
Биохимическая диагностика основана на выявлении аномальных дикарбоновых кислот и сопутствующих продуктов (глицина, карнитина) в моче. Глутаровая ацидурия и ацидурии органических кислот с разветвленными цепями сочетаются с дефицитом МАД. Следует подчеркнуть, что данные специфические показатели лучше всего выявляются при обострении, в то время как в стабильном состоянии наиболее достоверными тестами являются определение общего и эстерифицированного карнитина плазмы и аномальных показателей ацил-карнитина в плазме или в сухой капле крови. Определение скорости окисления жирных кислот в свежих лимфоцитах или культуре фибробластов используется для сужения диапазона поиска специфического дефекта, затем можно провести прямое ферментное исследование для уточнения диагноза. Большая часть дефектов выявляется в культуре фибробластов; тем не менее, определенные отклонения (такие как дефицит мышечной КПТ-1 и SCHAD) могут быть тканеспецифичными (Sim et al., 2002).
Упрощенная схема бета-окисления длинноцепочечных жирных кислот с указанием мест основных врожденных дефектов.
1 — первичный дефицит карнитина. 2 — карнитин ацилкарнитиновый шунт(карнитин-пальмитоилтрансфераза I и II, карнитин транслоказа).
3 — дегидротаза длинно- и сверхдлинноцепочечного ацил кофермента A (LCAD/VLCAD). 4 — система переноса электронов (МАД, глутаровая ацидурия II типа).
5 — дегидрогеназа длинноцепочечного 3 - гидрокси-ацил-кофермента A (LCHAD). 6 — трифункциональный белок.
б) Генетические изменения. Данные нарушения встречаются часто, но особенно распространены дефицит MCAD и СРТ-II. Все зарегистрированные нарушения наследуются аутосомно-рецессивным путем. У гетерозигот отклонений обычно не отмечается. Тем не менее, у гетерозиготных по гену LCHAD матерей, беременных больным плодом, к концу беременности может развиться острая жировая инфильтрация печени. Все пораженные гены были клонированы и зарегистрированы различные мутации, вызывающие заболевания. Преобладающие мутации связаны с MCAD, формой дефицита КПТ-II подростов и взрослых, и альфа-субъединицей ТФБ. Антенатальная диагностика возможна путем определения уровня ферментной активности в культуре амниоцитов или в образцах хорионических ворсин, более достоверна диагностика на основании молекулярного анализа. Высокая частота данных нарушений сочетается с высокой эффективностью лечения и/или профилактики, что является обоснованием для распространения скрининга новорожденных. Данный скрининг уже применяется во многих странах, особенно в отношении дефицита MCAD (Wilcken et al„ 2003).
в) Клинические проявления. У пациентов с нарушением окисления жирных кислот типичны клинические проявления, связанные с острым недостатком энергии, проявляющимся в любом возрасте, начиная с периода новорожденности до взрослого возраста (табл. 8.10). Тем не менее, новорожденные, младенцы и маленькие дети наиболее подвержены декомпенсации с связи с ограниченными запасами глюкозы. При рождении больные дети не способны справиться с энергетическими потребностями. У младенцев и маленьких детей длительное голодание и интеркуррентные инфекции являются наиболее частыми провоцирующими факторами. У детей старшего возраста декомпенсация может быть индуцирована инфекциями, лихорадкой и длительной физической нагрузкой.
Клинически острые проявления заболевания могут включать угрожающие жизни эпизоды гипокетоновой гипогликемии, кардиомиопатии и аритмии, кому и полиорганную недостаточность. Печеночная энцефалопатия с лактатацидозом и гипераммониемией часто путают с синдром Рея. Часто отмечаются миолиз и/или кардиомиопатия различной степени выраженности. В некоторых случаях стремительная неожиданная смерть может быть основанием для диагностики синдрома внезапной смерти младенцев. В большинстве случаев внезапное ухудшение состояния связано с гипокетоновой гипогликемией, лактатацидозом, гипераммониемией, признаками печеночной недостаточности и поражением мышц с повышением уровня креатинкиназы. Несмотря на коррекцию уровня глюкозы в крови, у некоторых пациентов может отмечаться персистирующая сонливость, припадки, дистонические движения или опистотонус вследствие сопутствующего отека головного мозга, что предполагает нейротоксическое действие повышенного уровня жирных кислот. Уровень смертности, особенно среди новорожденных, высокий. Среди выживших часто отмечаются бессимптомные промежуточные эпизоды. Иногда развивается хроническое поражение мышц и сердца или неврологические отклонения с припадками или задержкой развития.
Описанные неврологические последствия наблюдались при MCAD, заболевании, определяемом еще и как наименее выраженная форма нарушения окисления жирных кислот. У некоторых пациентов может отмечаться задержка развития, нарушения поведения, хронические припадки или двигательные нарушения, расцениваемые как детский церебральный паралич. По результатам небольшого количества нейровизуализационых исследований зарегистрированы следующие основные рентгенологические признаки: атрофия головного мозга, перивентрикулярная демиелинизация и повреждения базальных ганглиев (Brismar и Ozand, 1994а).
г) Специфические проявления. У новорожденных кроме острых проявлений также встречался аномальный органогенез с дисморфическими проявлениями. Кистозная дисплазия головного мозга и почек и полимикрогирия отмечаются в некоторых случаях дефицита MAD и КПТ-П (North et al., 1995). При дефиците SCAD основные неврологические отклонения такие как гипертония, гиперактивность, нистагм и задержка развития предполагают тяжелое нарушение созревания головного мозга, вероятно, вызванные специфическим нейротоксическим действием метаболитов короткоцепочечных жирных кислот (Bhala et al., 1995; Tein 2002).
Мышечная слабость и рабдомиолиз в результате нарушения энергопродукции являются основными проявлениями нарушения окисления жирных кислот. При таких заболеваниях, за исключением дефицита КПТ-1, описана хроническая мышечная слабость с миопатией и накоплением липидов. При дефиците КПТ-П описана тяжеля миопатия новорожденных без поражения органов (Land et al., 1995). Мышечная слабость в сочетании с прогрессирующей гипертрофической и/или дилатационной кардиомиопатией является характерным проявлением системного дефицита карнитина, распространенного в младенческом возрасте. Описаны случаи позднего начала заболевания во взрослом возрасте в сочетании с сердечной аритмией. Легкая форма MAD во взрослом и подростковом возрасте характеризуется прогрессирующей мышечной слабостью и непереносимостью нагрузок. Диагностика очень важна, так как у некоторых пациентов отмечается улучшение в ответ на применение рибофлавина.
При нарушениях, затрагивающих окисление длинноцепочечных жирных кислот (например КПТ-II, VLCAD, LCHAD, TFP, M/SCHAD и MAD), часто отмечаются эпизоды острого рабдомиолиза и серьезные осложнения. Чаще всего встречается легкая форма дефицита КПТ-II у подростков и взрослых. Заболевание характеризуется приступами рабдомиолиза без поражения других органов и без мышечной слабости в промежутках между приступами. Обострения провоцируются длительными нагрузками, голоданием, интеркурентными инфекциями или нахождением на холоде. Поиск дефектов окисления жирных кислот и особенно дефицита КПТ-П должен быть систематическим и включать диагностическое обследование всех пациентов, у которых впервые развился рабдомиолиз. Рецидивирующие эпизоды миолиза существенно замедляют течение заболевания у пациентов с дефицитом трифункционального протеина или LCAD, у которых в противном случае может развиться прогрессирующая аксональная нейропатия и пигментная ретинопатия (den Boer et al., 2003; Spiekerkoetter et al., 2004; Tyni et al., 2004).
д) Лечение. Основным методом лечения является предотвращение голодания и катаболических состояний. Во время обострения следует немедленно начать внутривенное введение глюкозы (10-12 мг/кг в минуту). Нет однозначных подтверждений, что ограничение потребления жира эффективно при дефиците MCAD. С другой стороны, для пациентов с нарушением метаболизма длинноцепочечных жирных кислот, может быть эффективна диета с низким содержанием жира и поддерживающим приемом триглицеридов. У пациентов с первичным дефицитом карнитина поддерживающее применение карнитина (100-300 мг/кг в сутки) является жизненно необходимым, так как в течение нескольких месяцев приводит к улучшению работы мышц и сердца. При всех других нарушениях вторичный дефицит карнитина обычно компенсируется при дозировке 50-100 мг/кг в сутки. У некоторых пациентов с легкими формами дефицита MAD и SCAD может отмечаться положительный эффект при поддерживающем применении рибофлавина (100 мг/кг в сутки). Доказано, что поддерживающее применение докозагексаеновой кислоты эффективно для предотвращения нейропатии и ретинопатиии у пациентов с LCHAD/TFP. Возможно предотвращение мышечной боли или непереносимости нагрузок путем предварительного приема кукурузного крахмала или триглицеридов с цепями средней длины (Gillingham et al., 2006; Ogier de Baulny и Superti-Furga, 2006).