Митохондриальная энцефаломиопатия у детей - MELAS-синдром
MELAS-синдром может не проявляться у детей в первые годы жизни, однако затем постепенно замедляется психомоторное развитие, характерен низкий рост.
Клиническая картина MELAS-синдрома характеризуется следующими признаками:
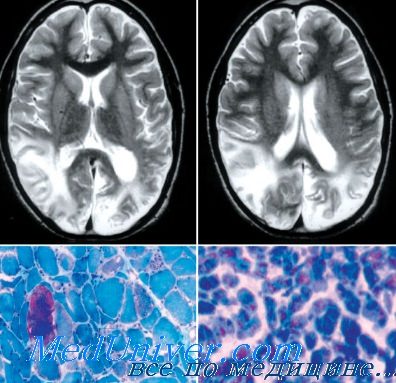
1) инсультоподобные эпизоды с локализацией поражения в большинстве случаев в задних отделах височной доли, в теменных и затылочных долях (признаки локального поражения мозга на КТ или МРТ);
2) лактат-ацидоз, феномен рваных красных волокон или их сочетание;
3) по крайней мере два из следующих симптомов: фокальные или генерализованные эпилептические приступы, деменция, рецидивирующая мигренозная головная боль и рвота.
В одном исследовании дебют до 15 лет отмечен у 62 % пациентов, наиболее распространенным проявлением заболевания служила гемианопсия или кортикальная слепота. Часто отмечается повышение уровня белка в СМЖ. Мутация MELAS 3243 может ассоциироваться с различными комбинациями симптомов: непереносимость физической нагрузки, миопатия, офтальмоплегия, пигментная ретинопатия, гипертрофическая или дилатационная кардиомиопатия, нарушение сердечной проводимости, глухота, эндокринопатия (сахарный диабет) и дисфункция проксимальных отделов почечных канальцев. MELAS-синдром — прогрессирующее заболевание.
Имеются сообщения о развитии заболевания у сиблингов. При прогрессировании заболевания отмечаются инсультоподобные эпизоды, приводящие к деменции.
При однофотонной эмиссионной компьютерной томографии (ОФЭКТ) может определяться региональная гипоперфузия. Патоморфологические исследования иногда выявляют кортикальную атрофию в сочетании с инфарктоподобными очагами поражения в корковых и подкорковых структурах, кальцинаты в базальных ганглиях и расширение желудочков. Мышечная биопсия, как правило, демонстрирует феномен рваных красных волокон.
Скопления и патологические изменения митохондрий определяются в гладких мышечных клетках внутримышечных сосудов и артериол мозга, в эпителиальных клетках и кровеносных сосудах хориоидальных сплетений, что приводит к митохондриальной ангиопатии.
Биохимическое исследование мышц выявляет дефицит комплекса I у многих пациентов, однако в некоторых случаях зарегистрированы множественные нарушения, включая дефицит комплексов I, III и IV. Заболевание характеризуется материнским типом наследования; высокоспецифичной (хотя и не единственно возможной) для данного заболевания считается точечная митохондриальная мутация гена тРНК лейцина, локализующаяся у 80 % больных в нуклеотидной позиции 3243. Еще у 7,5 % пациентов — точечная мутация гена тРНК лейцина в нуклеотиде 3271. Третья мутация выявлена в нуклеотидной позиции 3252 гена тРНК лейцина.
Поскольку концентрация мутантных генов ниже в клетках крови, чем в мышечной ткани, для диагностики заболевания предпочтительно исследование мышц.
Прогноз у пациентов с полной клинической картиной MELAS-синдрома неблагоприятный. Схема терапии включает применение кортикостероидов и коэнзима Q10. В некоторых случаях снижение уровня лактата в крови за счет приема дихлороацетата привело к выраженному клиническому улучшению.