Нервная система при нарушениях обмена холестерина: мевалоновой ацидурии
а) Мевалоновая ацидурия. Мевалоновая ацидурия является врожденным нарушением биосинтеза холестерина, вызванным дефицитом мевалонаткиназы. Данное нарушение проявляется двумя нозологическими формами: повышенным уровнем IgD и периодической лихорадкой (HIDS) и классической мевалоновой ацидурией (МА). HIDS характеризуется рецидивирующими приступами лихорадки, которые обычно начинаются на первом году жизни и не сопровождаются явными неврологическими аномалиями.
С другой стороны, vевалоновая ацидурия (МА) обычно характеризуется задержкой психомоторного развития, атаксией и дисморфическими проявлениями. У пациентов также отмечаются приступы лихорадки. В настоящее время оба нарушения расцениваются как единое заболевание с общими клиническими и биохимическими изменениями (Simon et al., 2004; Waterham и Clayton, 2006).
1. Биохимические изменения и патогенез. Мевалонаткиназа, которая фосфорилирует мевалоновую кислоту, является центральным ферментом изопренового пути с основными конечными продуктами холестерином, долихолом и убихиноном, что определяет решающую роль пути для пролиферации и функционирования клеток. Соответствующие патогенетические роли накопления конечных продуктов и токсического действия накопления мевалоната неясны.
Любая из этих ролей представляется достаточной для объяснения возникновения полиорганного поражения и тератогенного действия, предполагаемого на основании высокой частоты выкидышей и мертворождений плодов с мальформациями в семьях с данными заболеваниями. В постнатальном периоде отмечается высокая восприимчивость мозжечка и сетчатки к окислительному стрессу и/или токсическому действию мевалоновой кислоты. Механизм аутовоспалительного процесса остается невыясненным.
2. Генетические изменения. Мевалоновая ацидурия (МА) и HIDS являются аутосомно-рецессивными заболеваниями, выявлено около 40 вызывающих заболевание мутаций, затрагивающих ген МК. Некоторые мутации, проявляются преимущественно фенотипом HIDS, а три другие встречаются относительно часто. Большинство пациентов являются гетерозиготами по двум различным мутациям, сочетанное воздействие которых на остаточную ферментную активность отчасти объясняет вариабельность фенотипов. Активность мевалонаткиназы проявляется в фибробластах, амниотических клетках, лейкоцитах и хорионических ворсинах.
Пренатальная диагностика проводилась на основании исследования образцов ворсин хориона на предмет ферментной активности и содержания мевалоновой кислоты в амниотической жидкости. Более достоверная диагностика возможна по результатам молекулярного тестирования (Waterham и Clayton, 2006).
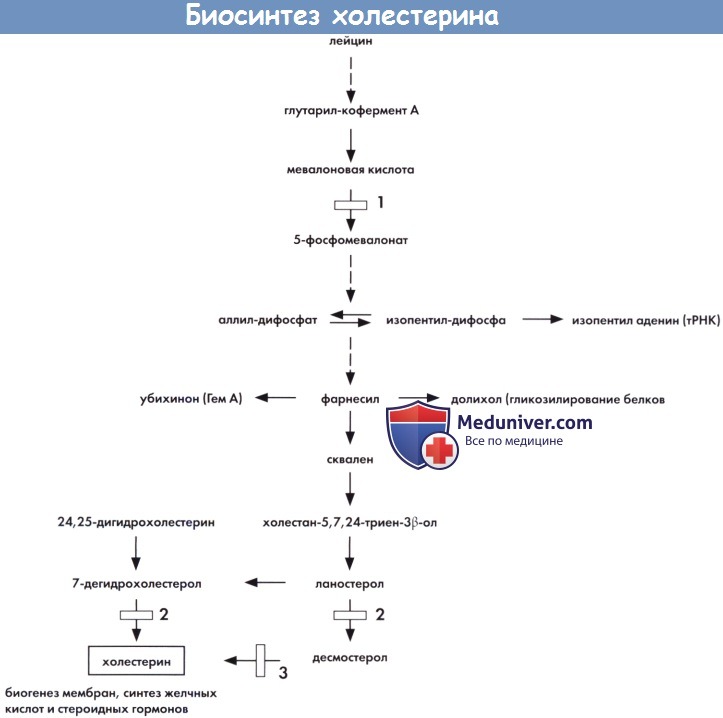
Схематичное изображение биосинтеза холестерина и основных взаимосвязей с другими путями метаболизма (см. текст).
1 — мевалонат киназа (мевалоновая ацидурия). 2 - 7-дегидрохолестерин дегидрогеназа (синдром Смита-Лемли-Опитца).
3 — редуктаза двойной связи С24-С25 (возможен дефект десмостеролозиса).
3. Нейропатология. При нейропатологическом исследовании одного пациента был выявлен агенез червя мозжечка, некроз гранулярных клеток и некроз нейронов коры головного мозга (Hoffman et al., 1993).
4. Клинические проявления. Заболевание может проявляться в младенческом или детском возрасте и характеризуется фенотипической гетерогенностью. Тяжелые формы могут приводить к смерти в раннем младенческом возрасте или даже во внутриутробном периоде. С другой стороны, при легких формах заболевания пациенты доживают до детского возраста с формированием различных нарушений. МА и HIDS обычно проявляются рецидивирующими эпизодами аутовоспаления, длящимися 3-5 дней. Обострения могут возникать до двух раз в месяц в младенческом возрасте и менее часты в позднем детском возрасте.
Обострения представлены выраженной лихорадкой, болью в животе, рвотой, диареей, лимфоаденопатиями, гепатоспленомегалией, артралгиями и кожной сыпью. Кроме того, у пациентов с МА отмечается задержка умственного развития различной степени выраженности. У большинства пациентов в течение двух лет развивается прогрессирующая атаксия, дизартрия и прогрессирующая мозжечковая атрофия. У всех пациентов отмечается мышечная гипотония и слабость. Часто встречается замедление скорости роста, анемия, перманентная гепатоспленомегалия и лимфоаденопатия. Легкий дисморфизм был зарегистрирован у 8 из 11 пациентов (Hoffman et al., 1993). Катаракта, увеит, прогрессирующая дистрофия сетчатки и атрофия зрительного нерва приводят к прогрессирующим ухудшениям зрения. У отдельных пациентов отмечались парциальные или генерализованные тонико-клонические припадки. При менее выраженной форме заболевания пациенты могут доживать до подросткового и даже до взрослого возраста с незначительной инвалидностью и гораздо менее острыми эпизодами лихорадки (Prietsch et al., 2003; Simon et al., 2004).
5. Биохимическая диагностика. Диагностика основана на определении уровня органических кислот для выявления накопления мевалоновой кислоты в жидкостях организма. Уровень мевалоновой кислоты обычно повышается в случае классического фенотипа, но встречается и при менее тяжелых формах заболевания. Постоянное повышение уровня IgD и/или IgA отмечается при HIDS, а при МА имеет непостоянный характер. Лучшим методом диагностики является определение активности ферментов в лейкоцитах или фибробластах, а также молекулярные исследования.
6. Лечение. В настоящее время эффективных методов лечения не существует. Поддерживающее лечение предположительной недостаточности конечных продуктов, таких как холестерин, желчные кислоты и убихинон, не привело к клиническому или биохимическому улучшению. Блокирование гиперпродукции мевалоната с помощью ингибитора HMG-кофермент А редуктазы (ловастатина) привело к тяжелым обострениям у двух пациентов с МА. Лучшим методом лечения считается интермиттирующее применение стероидов.
б) Другие нарушения метаболизма холестерина. Синдром Смита-Лемли-Опитца является комплексным пороком развития в результате нарушения биосинтеза холестерина в связи с блоком на уровне фермента 3-бета гидроксистерол D-7 редуктазы. Данное заболевание описано в отдельной статье на сайте.
Другие редкие мальформационные синдромы, вызванные дефектами биосинтеза холестерина, включают болезнь CHILD (врожденная гемидисплазия с ихтиозоформной эритродермией и дефектами конечностей), синдром Конради-Хюнермана (сцепленная с Х-хромосомой доминантная точечная эпифизарная дисплазия) и десмостеролоз (Haas et al, 2001).