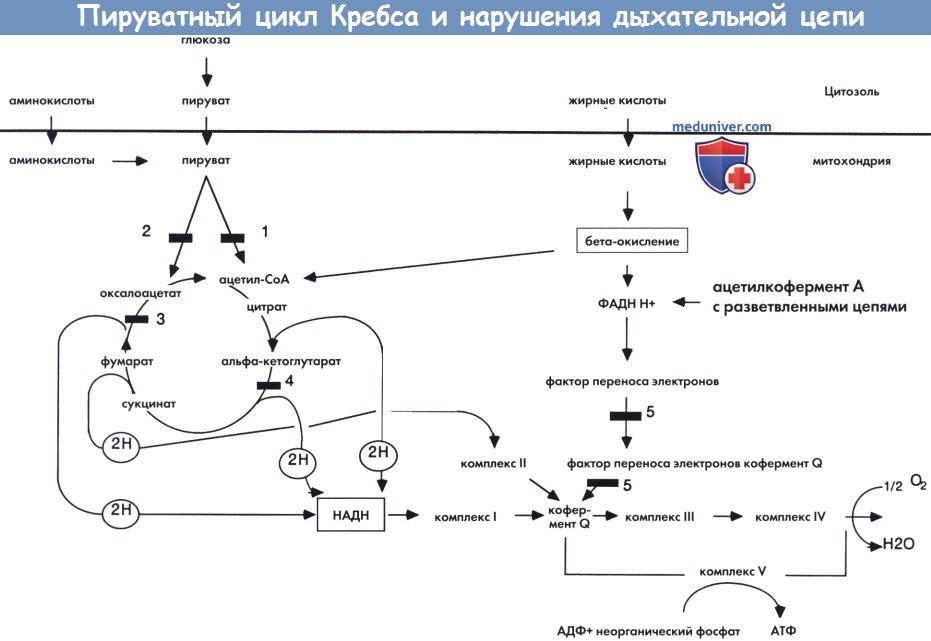
Нарушения цикла Кребса включают дефицит фумаразы, альфа- кетоглутаратдегидрогеназы и липоамиддегидрогеназы.
а) Дефицит дигидролипоамиддегидрогеназы (ДЛД). Дигидролипоамиддегидрогеназы (ДЛД) является компонентом ряда митохондриальных мультиферментов, включая пируват дегидрогеназу, кетоглутаратдегидрогеназу и дегидрогеназу альфа-кетокислот с разветвленными цепями (Cameron et al., 2006). Клинический фенотип дефицита ДЛД может варьировать от тяжелых неонатальных поражений до менее выраженных форм, проявляющихся в детском возрасте.
В младенческом возрасте заболевание проявляется интермиттирующим кетоацидозом, неврологическими изменениями, микроцефалией и синдромом Ли, которые приводят к задержке умственного развития, атаксии и гипотонии. Менее тяжелые формы, проявляющиеся в детском возрасте, характеризуются эпизодической рвотой, кетоацидозом, печеночной недостаточностью, гипотонией и непереносимостью нагрузок в промежутках между приступами у небольшого количества пациентов.
С биологической точки зрения периоды декомпенсации проявляются гиперлактацидемией, сопровождаемой повышением экскреции альфа-кетоглутаровой кислоты с мочой и (в качестве менее постоянного признака) повышением уровня аминокислот с разветвленной цепью в плазме. При нейровизуализации выявляются различные изменения от неспецифических повреждений перивентрикулярного белого вещества до признаков синдрома Ли с поражением базальных ганглиев. Зарегистрированы мутации гена ДЛД, типичная мутация отмечается среди потомков евреев ашкенази (229G >С в экзоне 9).
б) Дефицит фумаразы. Дефицит фумаразы был зарегистрирован у небольшого количества детей с тяжелыми неврологическими отклонениями (Kerrigan et al., 2000). Клинический фенотип в младенческом возрасте характеризуется выраженной задержкой развития и гипотонией, припадками, дисморфизмом лица, нарушением роста, относительной микроцефалией и смертью с первые годы жизни.
Гиперлактацидемия может быть слабовыраженной, но при этом отмечается экскреция фумаровой кислоты с мочой. По результатам МРТ головного мозга изменения могут иметь выраженный характер и сопровождаться вентрикуломегалией, полимикрогирией, агенезом мозолистого тела, снижением объема белого вещества и относительной гипоплазией ствола мозга. При патологоанатомическом исследовании была выявлена гипомиелинизация и гетеротопия, локализованная в мозжечке, затылочной и теменной областях (Gellera et al., 1990). Нарушения затрагивают как цитоплазматические, так и митохондриальные изоформы и наследуются аутосомно-рецессивным путем (Deschauer et al., 2006).
в) Дефицит альфа-кетолутарата. Дефицит альфа-кетоглутарата был описан у небольшого количества новорожденных и младенцев (Bonnefont et al., 1992, Surrendran et al., 2002). Клинические проявления напоминают дефицит ПДГ, начинающийся в раннем возрасте, или митохондриальные энцефаломиопатии. Легкая гиперлактатемия сопровождается аномальным уровнем альфа-кетоглутаровой кислоты в моче в сочетании с повышенным уровнем экскреции промежуточных продуктов цикла Кребса или без данного проявления. У всех пациентов отмечалась атрофия коры и в некоторых случаях некроз стриатума.
Пируватный цикл Кребса и нарушения дыхательной цепи.
1 — пируват дегидрогеназа. 2 — пируват карбоксилаза. 3 — фумараза.
4 — альфа- кетоглутаратдегидрогеназа. 5 — фактор переноса электронов и фактор переноса электронов кофермент Q дегидрогеназа.