Описание наследственных нарушений каждого этапа метаболизма витамина В12 (кобаламина) в новом свете представило патогенез дефицита кобаламина и фолата (Hall, 1990). Клинические проявления приблизительно одинаковы при всех врожденных нарушениях метаболизма кобаламина, за исключением изолированного дефицита аденозилкобаламина, проявления которого идентичны симптомам метилмалоновой ацидурии (Rosenblatt и Whitehead, 1999; Surtees, 2001; Whitehead, 2006).
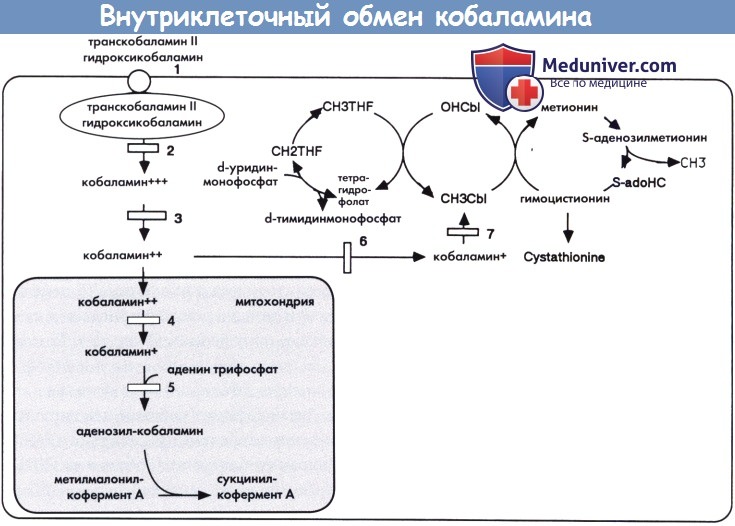
а) Биохимические изменения. Понятие «кобаламин» объединяет группу веществ, выполняющих функцию коферментов в двух клеточных реакциях человека: образовании метионина из гомоцистеина с метилкобаламином в качестве кофермента и превращении метилмалонил-кофермента А в сукцинил-кофермент А с аденозилкобаламином в качестве кофермента. Кобаламин, поступающий с пищей, преимущественно с животными белками, абсорбируется с помощью фактора Касла, затем поступает в кровоток в комплексе с транскобаламином-II после захвата клетками с помощью специфического поверхностного рецептора.
Внутри клетки кобаламин подвергается серии модификаций в результате которых образуется метилкобаламин и аденозилкобаламин. Дефицит кобаламина может быть классифицирован на четыре группы: дети, рожденные матерями с дефицитом кобаламина, дефицит системы абсорбции кобаламина, дефект транспорта витамина и нарушения внутриклеточной утилизации. Четвертая группа соответствует ряду дополнительных групп, обозначаемых от CblA до CblH, каждая из которых связана с специфическим ферментным блоком внутриклеточного метаболизма. В зависимости от участка ферментного дефекта, у пациентов отмечается метилмалоновая ацидурия или гомоцистинурия или их сочетание.
Уровень кобаламина в плазме остается нормальным при всех, за исключением одного (CblF), внутриклеточных дефектах метаболизма кобаламина, а также при дефекте транскобаламина II. С другой стороны, при врожденном гиповитаминозе и дефектах абсорбции отмечается низкий уровень кобаламина в плазме.
Дефицит транскобаламина II (1), кобаламина F (2) и кобаламина С или кобаламина D (3) приводят к нарушению синтеза кофакторов аденозил- и метил-кобаламина, в результате чего возникает комбинированная метилмалоновая ацидурия и гомоцистинурия в сочетании с гипометионинемией. Дефицит кобаламина А (4) и кобаламина В (5) приводят к нарушению синтеза только аденозил-кобаламина, в результате чего возникает изолированная метилмалоновая ацидурия. Дефицит кобаламина Е (6) и кобаламина G (7) приводит к нарушению синтеза только метил-кобаламина, в результате чего возникает гомоцистинурия и гипометионинемия (дефицит кобаламина А, В, С, D, Е, F и G является различными дополнительными группами метилмалоновой ацидурии: см. текст).
б) Генетические изменения. Все нарушения внутриклеточного метаболизма кобаламина наследуются аутосомно- рецессивным путем. Пренатальная диагностика может проводиться путем исследования амниотической жидкости и органических кислот. Дальнейшее подтверждение диагноза проводится на основании измерения количества определенных ферментов в культуре хорионических ворсин или с помощью молекулярного анализа, когда известно о наличии у пациента обеих мутаций (Morel et al., 2005).
в) Патофизиология. Многие неврологические, гематологические и биохимические проявления типичны для приобретенного дефицита кобаламина и фолата, врожденных дефектов абсорбции или транспорта кобаламина и фолата и внутриклеточных нарушений метаболизма кобаламина и фолата. Все виды нарушений приводят к дефициту метионинсинтетазы, играющей ключевую роль в патогенезе нарушений метаболизма кобаламина и фолата (Hall, 1990; Weir и Scott, 1995). Дефицит метионинсинтетазы обладает несколькими предположительными вторичными эффектами, включающими накопление метилтетрагидрофолата (СН3ТГФ) (теория фолатной ловушки), что препятствует ее участию в синтезе кофермента фолата, который необходим для синтеза ДНК и гемопоэза.
Дефицит метионинсинтетазы также приводит к нарушению синтеза метионина и аденозилметионина, который является основным донором метиловых остатков для метилирования многих субстратов, таких как нейротрансмиттеры, основный белок миелина и фосфолипиды плазматических мембран. Считается, что данное нарушение метилирования является причиной подострой сочетанной дегенерации спинного мозга, описанного как при приобретенном, так и врожденном дефекте. Другие факторы могут нарушать структуру и функцию центральной нервной системы при приобретенном и врожденном нарушении метаболизма кобаламина, например, токсическое действие накопленного гомоцистеина может стать причиной сосудистых повреждений и тромбоэмболии, и накопления метилмалонил-кофермента А и пропионил-кофермента А в связи с нарушением активности мутазы.
г) Клинические проявления:
1. Врожденный дефицит витамина В12. Дефицит кобаламина у детей на грудном вскармливании у матерей с субклинической пернициозной анемией или находящихся на строгой вегетарианской диете без достаточного поступления витамина В12 может привести к неврологическому регрессу, аномальным движениям и коме в сочетании с мегалобластной анемией. Введение витамина В12 приводит к быстрому улучшению неврологического статуса, но сохраняется задержка дальнейшего развития (Gutierrez-Aguilar et al., 2005).
2. Дефицит абсорбции и транспорта кобаламина. Нарушения абсорбции и транспорта кобаламина являются причиной синдрома дефицита кобаламина, характеризующегося прогрессирующим течением с началом в возрасте от 1 месяца до нескольких лет. Обычно первые симптомы связаны с желудочно-кишечным трактом, а также включают плохую прибавку в весе, мышечную слабость, вялость и мегалобластную анемию. Несколько месяцев спустя развиваются периферическая нейропатия, миелопатия и энцефалопатия с заметной отсрочкой развития. Нарушения абсорбции связаны с дефицитом фактора Касла или дефектом кубилинамниотического комплекса, который действует в качестве рецептора комплекса IF-кубилин (синдром Имеслунда-Гресбека) (Fyfe et al., 2004).
Дефект транспорта связан с дефицитом транскобаламина II, дефицит которого даже при лечении, может привести к тяжелым неврологическим нарушениям (Monagle и Tauro, 1995).
3. Нарушение внутриклеточного метаболизма. Внутриклеточный дефицит кобаламина, нарушающий синтез только метилкобаламина (кобаламин E/G/F) или метил- и аденозилкобаламина (кобаламин С/D) отмечается при трех основных типах заболевания. Чаще всего встречается тяжелая форма заболевания с началом в раннем возрасте, описанная у новорожденных и младенцев в возрасте до 3-х месяцев. Симптомы включают прогрессирующую сонливость, гипотонию, аномальные движения и/или припадки в сочетании с панцитопенией и мегалобластной анемией. У некоторых пациентов также отмечается полиорганная недостаточность, включающая почечную недостаточность с гемолитическим уремическим синдромом, кардиомиопатию и интерстициальную пневмонию. Поражение сетчатки с гранулярной депигментацией макулы и дальнейшим периферическим пигментным ретинитом часто является ранним признаком дефицита кобаламина С. Сообщающаяся гидроцефалия может быть дальнейшим осложнением.
У небольшого количества пациентов заболевание проявляется в детском возрасте в виде прогрессирующей неврологической деградации, микроцефалии, эпизодических припадков и мегалобластной анемии, которая обычно является основанием для постановки диагноза. При отсутствии лечения острая неврологическая деградация может сочетаться с признаками и симптомами, напоминающими подострую дегенерацию спинного мозга. В редких случаях развития заболевания у подростков и взрослых, отмечалась сходная подострая дегенерация спинного мозга, которой предшествовала острая деградация интеллектуальных функций и иногда поведенческие нарушения. Среди пациентов старшего возраста мегалобластная анемия может быть едва заметной, а пограничный макроцитоз следует рассматривать особенно тщательно (Ogier de Baulny et al., 1998; Roze et al., 2003). Fla KT и MPT выявляется атрофия мозга и/или демиелинизация (Rossi et al., 2001).
д) Лечение нарушения обмена витамина В12. Следует систематически исследовать чувствительность к поддерживающей терапии витамином В12. В зависимости от имеющегося дефекта выбирается использование парентерального или перорального пути введения, фармакологических или минимальных доз и гидроксикобаламина или естественных субстратов кобаламина (метил-, аденозилкобаламина). При нарушении транспорта и внутриклеточных дефектах может потребоваться пополнение запасов метионина путем перорального введения бетаина или дополнительного применения метионина. На основании метаболического пути бетаина предполагается, что дополнительное применение фолиевой кислоты может быть полезным при длительном лечении бетаином. В связи с тем, что эндогенный синтез карнитина зависит от метионина, при состояниях, сопровождающихся нарушением синтеза метионина, может быть эффективно поддерживающее применение карнитина. Логично соблюдение диеты с нормальным содержанием белка и отказ от повышенного потребления белка (Ogier de Baulny et al., 1998).