Дыхательная цепь является основной биохимической системой, используемой для получения аэробной энергии всеми эукариотическими организмами. У человека нарушение данных процессов является причиной множества различных митохондриальных болезней, при которых обычным проявлением является нарушение продукции АТФ наряду с недостаточным снабжением энергией одной или нескольких тканей или органов. Эффект варьирует в различных тканях в соответствии с зависимостью от окислительного фосфорилирования.
Мышечная ткань в целом более чувствительна к снижению поступления АТФ, что приводит к появлению различных симптомов, таких как птоз, офтальмоплегия, мышечная слабость, непереносимость нагрузки и кардиомиопатия. Центральная нервная система также весьма чувствительна к дефициту АТФ, приводящему к энцефалопатии, сенсоневральным изменениям и периферической нейропатии. В зависимости от распространенности и выраженности дефекта возможно поражение и других тканей. Чаще всего поражаются почки, печень и ткани желудочно-кишечного тракта.
Клинические проявления митохондриальных заболеваний чрезвычайно гетерогенны. Заболевания могут поражать единичные органы или ткани, но чаще всего проявляются мультисистемными отклонениями, среди которых синдромы, регистрируемые на протяжении нескольких лет (Zeviani и Di Donato 2004; DiMauro и Hirano 2005).
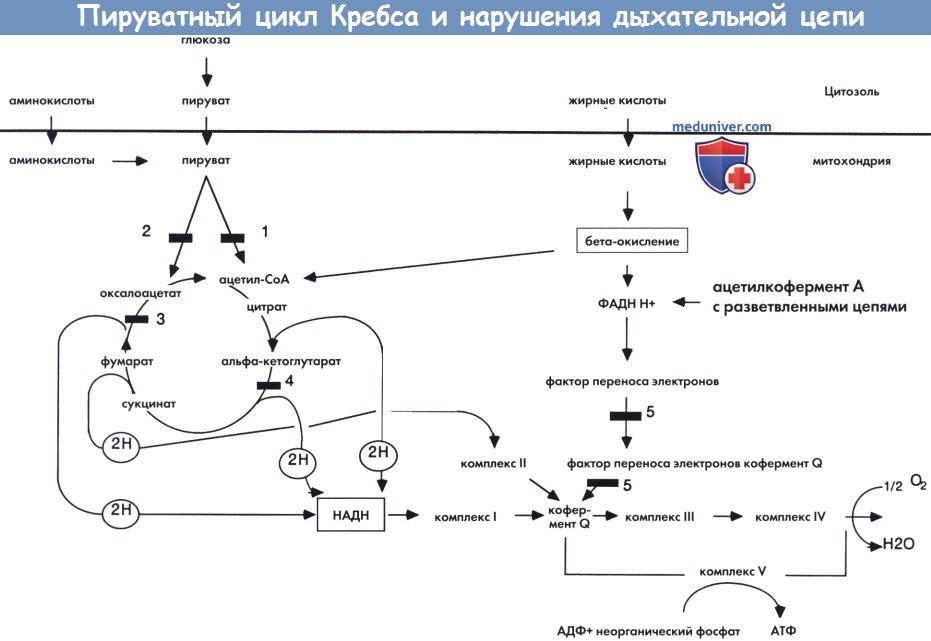
а) Функциональная структура дыхательной цепи. Митохондриальная дыхательная цепь представляет собой группу из пяти ферментных комплексов, встроенных во внутреннюю митохондриальную мембрану. Каждый комплекс состоит из множества полипептидных субъединиц и простетической группы. Кроме того, в состав дыхательной цепи входят два переносчика электронов, коэнзим Q10 и цитохром С. Дыхательная цепь организована таким образом, чтобы служить акцептором электронов от НАДН и ФАДН,, что снижает воздействие факторов, образующихся в процессе промежуточного метаболизма.
Электроны переносятся вдоль дыхательной цепи и соединяются с молекулярным кислородом с образованием воды. Одновременно высвобождающаяся при переходе энергия используется комплексами I, III и IV для переноса протонов (Н+) из митохондриального матрикса в межмембранное пространство. Комплекс V позволяет протонам вернуться в митохондриальный матрикс и использует высвобождаемую энергию для синтеза АТФ из АДФ и минерального фосфата (Chinnery и Schon, 2003; DiMauro и Schon, 2003).
Пируватный цикл Кребса и нарушения дыхательной цепи.
1 — пируват дегидрогеназа. 2 — пируват карбоксилаза. 3 — фумараза.
4 — альфа- кетоглутаратдегидрогеназа. 5 — фактор переноса электронов и фактор переноса электронов кофермент Q дегидрогеназа.
б) Генетические изменения. Приблизительно 90 белковых субъединиц, которые составляют пять комплексов, находятся под двойным генетическим контролем и включают белки, кодируемые ядерной ДНК (яДНК) или митохондриальной ДНК (мтДНК); исключением является комплекс II, который полностью кодируется яДНК. Человеческая мтДНК содержит гены 13 OXPHOS субъединицы (то есть дыхательную цепь, цепь переноса электронов), а также 22 транспортных РНК (тРНК) и две рибосомальных РНК (рРНК), необходимых для внутримитохондриального синтеза. Оставшиеся структурные белки кодируются яДНК, которая также кодирует сотни дополнительных факторов, необходимых для экспрессирования, транспорта и сборки белковых субъединиц, а также для синтеза, экспрессирования и стабилизации мтДНК.
Кроме того, митохондрии не являются статическими органоидами и другие факторы, кодируемые в ядре, регулируют их подвижность, деление и слияние. Установлено, что в общей сложности для работы дыхательной цепи необходимо около 1000 отдельных белков, в основном кодируемых яДНК, мутации которых потенциально являются причинами менделирующих наследственных заболеваний.
Следовательно, митохондриальные заболевания могут быть вызваны мутациями как мтДНК, так и яДНК. Тем не менее, неотличимые клинически заболевания могут быть вызваны отдельными мутациями разных генов в разных геномах. С другой стороны, одна специфическая мутация может приводить к формированию индивидуально варьирующих клинических симптомов.
в) Мутации мтДНК. Данные изменения включают точечные мутации и крупные реаранжировки. Точечные мутации впервые были описаны в генах, кодирующих различные тРНК, что приводило к широко распространенному нарушению синтеза белка. Такие мутации обычно гетероплазматические, наследуются от матери и сочетаются с различными мультисистемными нарушениями. Описаны точечные, приводящие к развитию заболевания, мутации гена 12S рРНК.
Точечные мутации в генах, кодирующих белки, поражают одну из кодируемых в митохондриях субъединиц комплекса I (НАДН дегидрогеназы), комплекса III (цитохрома b), комплекса IV (ЦОГ) или комплекса V (АТФазы 6). У пациентов отмечается склонность скорее к специфически мышечным изменениям, чем к мультисистемным нарушениям, и не выявляется наследование от матери. Тем не менее, ND5, кодирующий 5 субъединицу I комплекса, является участком возникновения множества точечных мутаций, сочетающихся с различными мультисистемными нарушениями.
Значительные перестановки включают уникальные делеции/дупликации мтДНК. Их размер индивидуален, но всегда приводит к делеции нескольких генов, кодирующих белки и тРНК. Данные мутации имеют склонность к спонтанному возникновению и являются гетероплазматическими. Тем не менее, в некоторых случаях возможно наследование от матери с различным фенотипическим экспрессированием у больной женщины и ее потомства.
г) Мутации ядерных генов. Данные изменения включают мутации генов, кодирующих субъединицы комплексов, вспомогательные белки, белковые факторы, необходимые для внутригеномной передачи сигнала, и белковые факторы, необходимые для поддержания мембраны и подвижности митохондрий. Данные мутации являются причиной менделевских аутосомно-рецессивных, доминантных или сцепленных с Х-хромосомой наследственных заболеваний.
Все в большем количестве выявляются мутации генов, кодирующих субъединицы дыхательной цепи. Различные мутации генов, кодирующих субъединицы комплекса I (NDUFS и NDUFV) и комплекса II (SDHA), в основном являются причиной синдрома Ли или подобного синдрома. Выявлены мутации генов (АРТХ, PDSS2, COQ2), кодирующих ферменты, участвующие в биосинтетическом пути кофермента Q10 (Lopez et al., 2006).
Единственный известный ядерный ген, изменения которого принимают участие в формировании дефицита комплекса III, кодирует дополнительный белок, необходимый для соединения единиц. Мутации являются причиной ранних и тяжелых мультисистемных заболеваний, впервые описанных среди жителей Турции; данное заболевание в Финляндии, где отмечается его высокая частота, также называют GRACILE синдром (задержка роста, аминоацидурия, холестаз, избыток железа, лактатацидоз и ранняя смерть). Мутации гена SURF1, отвечающего за сборку ЦОГ, часто сочетаются с синдромом Ли, вызванным дефицитом комплекса VI. Мутации других четырех генов (SCO1, SCO2, СОХ10, СОХ15), отвечающих за сборку ЦОГ, были описаны у пациентов, страдающих энцефалопатией, подобной синдрому Ли, и другими поражениями внутренних органов.
Отдельная форма синдрома Ли с дефицитом ЦОГ, вызванного мутациями гена LRPPRC, была описана в семьях франкоканадцев.
У пациентов с дефицитом мышечной ЦОГ и этилмалоновой ацидурией отмечалась мутация гена ETHE1. В конечном счете изменение гена АТР12 приводит к повреждению сборки комплекса V и становится причиной смертельной энцефалопатии.
в) Мутации в генах, кодирующих факторы внутригеномной передачи сигнала. Данные менделевские заболевания с качественными (делеции) или количественными (деплеции) повреждениями мтДНК являются результатом дисбаланса поступления деоксирибонуклеотидов в митохондрии (Alberio et al., 2007). В патогенезе синдромов митохондриальной деплеции (СМД) участвуют пять генов, ген тимидинкиназы (ТК2) при миопатических формах, ген β-субъединицы АДФ-образующей сукцинил кофермент А синтетазы (SUCLA2) при энцефалопатических формах и ген каталитической субъединицы полимеразы мтДНК (POLG) и MPV17 при гепатоцеребральных формах.
Описано четыре гена в поражениях с множественными делециями или деплециями. Мутации полимеразной части гена POLG встречаются при гетерогенной группе синдромов (Horvath et al., 2006). Это наиболее частая причина прогрессирующей наружной офтальмоплегии, кроме того они сочетаются с большим количеством клинических проявлений, включая синдром Альперса (заболевание, встречающееся в детском возрасте и характеризующееся печеночной недостаточностью и поражением серого вещества) (Gordon, 2006), SANDO (сенсорная атаксическая нейропатия, дизартрия, офтальмоплегия), глухоту, гипогонадизм, припадки, миоклонус, нарушение моторики желудочно-кишечного тракта, имеющее сходство с синдромом MNGIE (митохондриальной нейрогастроинтестинальной энцефаломиеолопатией) и, что важно, паркинсонизм.
Ген, ответственный за синдром MNGIE, кодирует фермент тимидинфосфорилазу (ТФ) (Hirano et al., 2005) и к настоящему моменту уже описано множество его мутаций. Два других гена являются причиной аутосомно-доминантной прогрессирующей внешней офтальмоплегии и множественных делеций ДНК в мышцах, в частности, ген ANT1, кодирующий изоформу транспортера адниннуклеотида, и ген twinkle, кодирующий митохондриальную хеликазу.
Мутации дополнительных ядерных генов являются причиной дефекта синтеза митохондриальных белков. Ген EFG1, кодирующий фактор элонгации, и ген MRPS16, кодирующий митохондриальную рибосомальную субъединицу, связаны с развитием раннего гепатоцеребрального синдрома. Ген PUS1, кодирующий митохондриальный фермент псевдоуридинсинтетазу, связан с миопатией, лактатацидозом и сидеробластной анемией (MI.ASA) (Bykhovskaya et al., 2004).
Хотя бы некоторые белки необходимы для того, что можно назвать «поддержанием» митохондрий. К неполностью изученным митохондриальных дисфункциям относится синдром Барта (сцепленная с Х-хромосомой миопатия, кардиомиопатия, нейтропения и 3-метилглутаконовая ацидурия) с низким уровнем кардиолипинов вследствие мутаций гена tafazzin. Кардиолипин (основной фосфолипидный компонент внутренней митохондриальной мембраны) регулирует активность нескольких комплексов дыхательной цепи. Дефекты белка, необходимого для подвижности митохондрий, были описаны при аутосомно-доминантной наследственной спастической параплегии с мутациями одного гена кинезина (KIFA). Дефекты белков, необходимых для митохондриального синтеза, были зарегистрированы при аутосомно-доминантной атрофии зрительного нерва (ОРА-1) и при аутосомно-доминантном варианте синдрома Шарко-Мари-Тута 2А типа с дефектом митофуссина 2 (MNF2).
Несмотря на возрастающее количество информации следует подчеркнуть, что молекулярные исследования все еще не позволяют идентифицировать генетический дефект у 80-90% пациентов детского возраста, у которых в соответствии с клиническими, биологическими и морфологическими признаками имеются нарушения дыхательной цепи.
г) Биологические проявления и диагностические тесты. В педиатрической популяции большая часть (90%) доказанных дефектов дыхательной цепи проявляется повышением уровня лактата в крови, мочевины — в спинномозговой жидкости, часто в сочетании с повышением соотношения лактат: пируват, что свидетельствует о изменении клеточного окислительно-восстановительного статуса. МР спектроскопия головного мозга является неинвазивным и эффективным методом оценки накопления лактата. Кроме того, она позволяет легко диагностировать редко встречающийся дефицит комплекса II по наличию аномального пика сукцината (Lin et al., 2003; Brockmann et al., 2005). У некоторых пациентов в состоянии покоя лактатацидоз не развивается, это зависит от выраженности энергетических нарушений, поражения органов и является следствием первичного дефекта скорости окисления. В некоторых случаях лактатацидоз может быть спровоцирован инфекцией или нагрузочным тестом с глюкозой.
У таких пациентов (обычно с непереносимостью нагрузки) может развиться тяжелая гиперлактацидемия после нетяжелых физических нагрузок. Тем не менее, лактатацидоз не является необходимым признаком заболевания, особенно в позднем детском или подростковом возрасте. Помимо обычной лактатурии, в редких случаях отмечается аномальная экскреция с мочой промежуточных продуктов цикла Кребса, таких как фумарат, альфа-кетоглутарат, сукцинат или 3-метилглутаконовая кислота. Легкая метилмалоновая ацидурия выявлялась в сочетании с мутацией гена SUCLA2, отвечающего за деплецию мтДНК (Carrozzo et al, 2007; Ostergaard et al., 2007). Этилмалоновая ацидурия является признаком этилмалоновой энцефалопатии (состояния, включающего выраженный дефицит комплекса IV в мышцах). Высокие уровни тимидина, деоксиуридина и урацила в крови и моче свидетельствуют о синдроме MNGIE с дефицитом тимидинфосфарилазы. Дефицит карнитина в сыворотке и мышцах может отражать недостаточность окисления жирных кислот. Уровень креатинкиназы (обычно умеренно повышенный) может быть высоким при миопатических формах синдрома деплеции мтДНК (СДМ).
МР спектроскопия может быть полезна у детей старше восьми лет, подростков и взрослых. Оценка соотношения фосфокреатина и минерального фосфора позволяет выявить дефект синтеза АТФ при мышечной нагрузке.
У пациентов с явно выраженным клиническим синдромом диагноз может подтверждаться простым молекулярным генетическим тестированием образцов ДНК. Образцами служат синдром MNGIE (ТР), младенческая энцефалогепатопатия (DGUOK) и младенческая энцефалотубулопатия (DCS1L). Обследование остальных пациентов является более сложным, особенно в связи с тем, что патогенетические мутации могут не определяться в анализе крови. При подозрении на митохондриальную болезнь следует проводить биопсию мышц (Chinnery и Schon, 2003).
Гистохимическое исследование с помощью окраски по Гомори, окраски цитохром С оксидазы и сукцинат-дегидрогеназы (СДГ) позволяет выявить митохондриальную пролиферацию и мозаицизм, что предполагает мутации мтДНК и дефицит комплексов II и IV.
Биохимические исследования включают два основных неисключительных теста с использованием обогащенной митохондриями фракции свежих тканей при полярографических исследованиях или с использованием замороженных тканей для анализа индивидуальной активности ферментов дыхательной цепи с помощью спектрофотометрии. Для исследований чаще всего используются образцы скелетных мышц. Тем не менее, возможно проведение специфических исследований на фибробластах, клетках крови, печени, почек и головного мозга. Теоретически для исследования подходит любая клинически пораженная ткань, т. е. при гепатоэнцефалопатии рекомендовано исследование образца печени. В случае сомнительных результатов биохимического анализа при выраженных клинических проявлениях следует провести исследование нескольких тканей.
При структурном подходе молекулярное исследование следует начинать с саузерн-блоттинга мтДНК для поиска перегруппировок мтДНК и серии аллель-специфических тестов для поиска часто встречающихся точечных мутаций. Дальнейший молекулярный скрининг с учетом клинического фенотипа, семейного анамнеза и результатов биохимических тестов, должен способствовать определению возрастающего количества заболеваний, что обеспечит более точное генетическое консультирование (Chinnery и Schon, 2003; DiMauro et al., 2004b). Основной проблемой точной диагностики митохондриальных заболеваний является отсутствие определенных биомаркеров, которые характеризуют заболевание у всех пациентов. Для улучшения интерпретации были разработаны диагностические схемы для младенцев и детей с целью разграничить вероятность митохондриального заболевания у конкретного пациента на определенную, возможную, вероятную или маловероятную (Bernier et al., 2002, Wolf и Smeitink, 2002).
Видео урок цикл Кребса - кратко, понятно, простым языком