а) Классический синдром линейного невуса. В полной форме данный синдром включает кожный невус, неврологические нарушения и аномалии глаз. Возможно несколько типов патологии кожи. Типичным кожным проявлением является сальный невус Ядассона. Данная патология чаще всего затрагивает голову или лицо и может выявляться при рождении или в младенческом возрасте, или становиться заметной через несколько лет (Clancy et al., 1985).
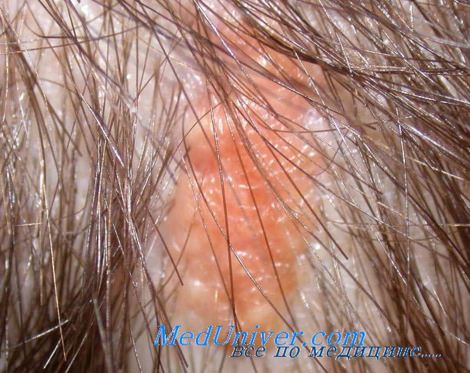
Сальный невус представляет собой несколько приподнятую желто-оранжевую гладкую бляшку линейной формы, которая граничит со средней линией лба, носа или губ и часто затрагивает кожу черепа. С течением времени образования темнеют и приобретают бородавчатую поверхность. В раннем возрасте выявляется главным образом акантоз и небольшое количество пигмента, а сальные железы обычно не выдаются. В более позднем возрасте сальные железы подвергаются пролиферации, а расположение апокринных желез в толще кожи может носить блуждающий характер.
Вторым типом невусов является бородавчатый невус, образованный из отдельных папул, несколько темнее окружающей кожи и с линейной организацией. В отличие от сальных невусов, они часто располагаются на теле и реже на лице, а гистологически представлены только акантозом и гиперкератозом с аномальной дермой (Prensky, 1987). Несмотря на то, что большая часть поражений кожи носит изолированный характер, предполагается, что оба типа невусов могут сочетаться с неврологическими отклонениями (Clancy et al., 1985).
Часто встречаются сочетанные поражения кожи, такие как ихтиоз, акантокератодермия, гемангиомы и пятна цвета «кофе с молоком», а линейный невус входит в состав других нейрокожных синдромов, сопровождающихся пролиферацией в коже и поражением подкожных тканей (см. ниже).
Неврологические изменения, сочетающиеся с обоими типами невусов (как в рамках синдрома Фойерштайна-Мимса), включают задержку умственного развития, припадки, паралич черепных нервов, гидроцефалию и асимметричную макроцефалию (van de Warrenburg et al., 1998). Припадки часто имеют парциальный характер, но в рамках одного исследования инфантильные спазмы отмечались у 4 из 11 пациентов (Vigevano et al., 1984).
Невус Ядассона
С точки зрения патологической анатомии, наиболее частым изменением со стороны головного мозга являются нарушения пролиферации и миграции (Prayson et al., 1999). Гемимегалэнцефалия встречается часто (Pavone et al., 1991) и может сопровождаться гемигипертрофией той же стороны лица и тела. У нескольких пациентов были выявлены гамартомные опухоли (Clancy et al., 1985). Также обнаруживаются деструктивные изменения, такие как порэнцефалия (Baker et al., 1987, Prensky, 1987), и имеющие сосудистое происхождение (артериальные аневризмы, артериовенозные мальформации и аномалии венозного возврата) (Dobyns and Garg 1991). Сходные изменения обнаруживаются в спинном мозге.
Другие аномалии центральной нервной системы включают кисты паутинной оболочки, субтотальную агенезию мозжечка и агенезию мозолистого тела в сочетании с мальформацией Денди-Уокера (Dodge и Dobyns, 1995).
Наиболее частой аномалией глаз являются дермоидные или эпидермоидные кисты конъюнктивы и колобомы радужки, сосудистой оболочки, сетчатки или зрительного нерва. Может отмечаться увеличение глазницы и большого крыла клиновидной кости на стороне невуса. Семейные случаи встречаются редко (Meschia et al., 1992).
На ранней стадии заболевания до появления видимых невусов диагностика может быть затруднена. Наиболее частой находкой на КТ является гипертрофия одного полушария (обычно на стороне невуса) с увеличением желудочка с той же стороны и пахигирия с понижением плотности белого вещества (Kruse et al., 1998). Такие изменения соответствуют изменениям в виде гемимегалэнцефалии или других нарушений миграции.
На ЭЭГ обычно обнаруживается пароксизмальная активность в области пораженного полушария, иногда с признаками гипсаритмии. Прогноз обычно неблагоприятный, но легкие случаи заболевания проявляются только припадками или небольшими нарушениями обучения. Описанный синдром носит спорадический характер, и причины его неизвестны. Относительно высокая частота опухолей позволяет предположить тесную взаимосвязь данного заболевания с факоматозами. Лечение носит симптоматический характер. У некоторых пациентов возможно удаление кожных изменений.
Сальной невус Ядассона
б) Родственные синдромы с пролиферацией кожи и поражением подкожных тканей:
1. Энцефалокраниокутанный липоматоз (ЭККЛ) и синдром с поражением глаз, мозга и кожи (ГМКС). Данные синдромы редки, тесно связаны, встречаются в основном спорадически и характеризуются микроцефалией, липодермоидами конъюнктивы, склеры или век и липоматозным отеком черепа или лица. Встречаются судороги и задержка умственного развития, на КТ возможны признаки атрофии головного мозга, кистозные участки и внутричерепная кальцификация. В рамках некоторых исследований (Fishman, 1987) было продемонстрировано, что мальформации головного мозга в виде порэнцефалии отмечаются во всех случаях, разрастания на веках—в большинстве случаев, а аномалии радужки — в двух случаях.
По результатам двух персональных и девяти опубликованных исследований были обобщены сведения о проявлениях со стороны головного мозга (Moog et al, 2005); данные проявления включали преимущественно микрогирию лобной коры и перивентрикулярные гетеротопии, агенезию мозолистого тела, гигантскую покрышку ствола мозга и отсутствие или гипоплазию червя или всего мозжечка. Данные состояния могут быть связаны с синдромом Деллемана (Pascual-Castroviejo et al., 2005b) и синдромом Протея (McCall et al., 1992).
2. Синдром Протея. Определение относится к множественному гамартомному синдрому, включающему частичный гигантизм, асимметрию конечностей, линейный бородавчатый и внутрикожный невус, шагреневые бляшки и различные сочетания лимфоангиоматоза, гемангиом, микроцефалии и гиперостозов. Гиперплазия кожи стоп (так называемая «мокасиновая стопа») является частым и значимым диагностическим признаком (Nguyen et al., 2004). Возможны эпибульбарные дермоидные кисты, колобомы, глаукома и отслойка сетчатки. Часто встречается гемимегалэнцефалия (Griffiths et al., 1994). Тератомы, являющиеся проявлением синдрома, могут достигать огромных размеров, как в случае Джозефа Мерика, так называемого «человека-слона». В случае данного заболевания до сих пор часто ошибочно диагностируется нейрофиброматоз.
Само определение синдрома говорит о возможности разнообразных проявлений. Большая часть выявленных к настоящему времени случаев носили спорадический характер. Мозаицизм соматических клеток, приводящий к летальному исходу в гомозиготном состоянии, может обуславливать большую часть проявлений синдрома, но является недоказанным (Cohen, 2005). Некоторые случаи сопровождаются мутацией гена PTEN (супрессора опухолей) (Thiffaut et al., 2004). Возможно выявление опухолей внутренних органов (Gordon et al., 1995). Диагноз заболевания затруднен, так как критерии синдрома четко не определены. Среди 205 зарегистрированных случаев только 97 (43%) отвечали четким критериям (Turner et al., 2004). Случаи заболевания без четких критериев иногда называют «Протей-подобным синдромом» (Zhou et al., 2000).
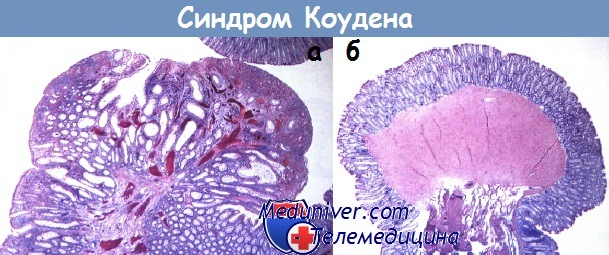
а - Синдром Коудена. Микроскопическая картина типичной гамартомы слизистой оболочки.
Пролиферация ветвистых фокально расширенных желез с умеренным количеством воспаленной стромы, которая может содержать пучки гладких мышц, фокальная эрозированная поверхность.
б - Второй тип гамартом при болезни Коудена — эквивалент лейомиомы мышечной пластинки слизистой оболочки.
в) Другие синдромы с пролиферацией тканей. Синдром Каудена характеризуется сочетанием различных поражений кожи, особенно в виде роговых папул на лице и других аномалий, и гамартом, которые могут включать полипы кишечника. В детском возрасте клинические проявления включают макроцефалию, складчатый язык и задержку умственного развития от легкой до умеренной степени выраженности, в то время как опухоли кожи и другие признаки появляются только в пожилом возрасте (Hanssen и Fryns 1995). Наиболее четким неврологическим проявлением является гамартомная гиперплазия мозжечка при болезни Лермитт-Дуклос (также вызванной мутацией гена PTEN), в действительности являющимся частью спектра синдрома Коудена (Perez-Nunez et al., 2004).
Синдром Баннаяна-Райли-Рувалькаба в некоторых случаях сопровождается мутацией гена PTEN (Hendricks et al., 2003). Полная форма синдрома представлена макроцефалией, полипами кишечника, липомами, ангиомами, пигментацией кожи и слизистых, отставанием в развитии или задержкой умственного развития.