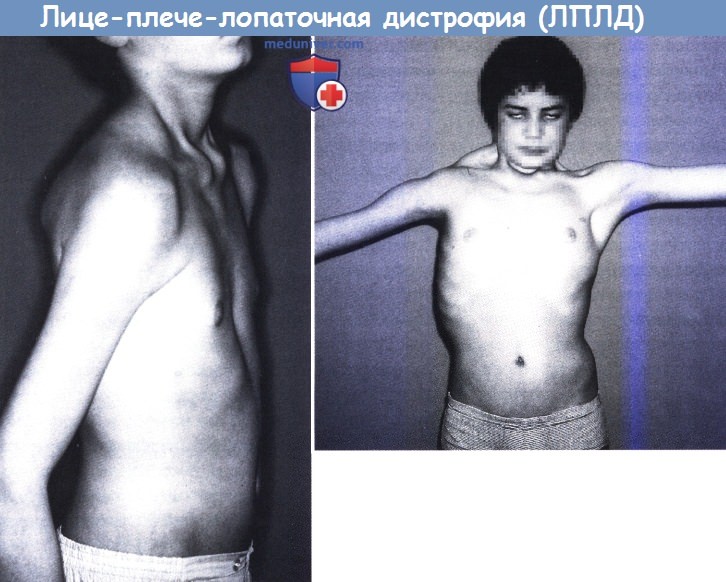
Лице-плече-лопаточная дистрофия (ЛПЛД) — это аутосомно-доминантное заболевание, характеризующееся специфической прогрессирующей мышечной слабостью, поражающей мышцы лица, мышцы — стабилизаторы лопатки, мышцы плеча, голени (малоберцовые мышцы) и пояса нижних конечностей (Tawil et al., 1998). Часто встречается асимметрия мышечной силы конечностей и/или плеч (Kilmer et al., 1995).
Обычно первые симптомы — это истощение плечевого пояса, крыловидные лопатки, поражение мышц лица — неспособность поджать губы, плотно сомкнуть глаза, слабость скуловой мышцы и поперечная улыбка. Может развиваться более или менее тяжелое поражение бицепсов и трицепсов плеч, и разнообразные поражения нижних конечностей, со слабостью малоберцовых мышц, со слабостью мышц пояса нижних конечностей или без. Дельтовидные мышцы остаются относительно сохранными до поздних стадий болезни. Функция дыхания обычно не страдает (Tawil и Griggs, 1997).
Мышечная слабость прогрессирует неравномерно, могут наблюдаться периоды явной стабилизации. Обычно у больных с лице-плече-лопаточной дистрофией (ЛПЛД) симптоматика развивается во второй декаде жизни, но возраст начала болезни вариабелен. Более чем у 90% пациентов симптомы заболевания манифестируют в возрасте около 20 лет; у некоторых симптоматика не развивается на протяжении всей жизни. В отдельных семьях может отмечаться ранняя манифестация болезни (Bailey et al., 1986), описано начало болезни в младенческом возрасте или раннем детстве, проявившееся слабостью мышц лица.
Лице-плече-лопаточная дистрофия у 13-летнего мальчика. Выраженное поражение плечевого пояса, больше справа.
Невозможность полностью закрыть глаза (вверху). Тот же пациент (слева), отмечается атрофия мышц шеи, амиотрофия мышц плеча и крыловидная лопатка.
Дополнительные симптомы включают в себя бессимптомную ретинальную ангиопатию (телеангиэктазии и микроаневризмы), выявляемые при флюоресцентной ангиографии у 40-60% пациентов и сенсоневральную потерю восприятия высоких частот на аудиограмме примерно у 60% (Padberg et al., 1995). Уровень креатинкиназы (КК) сыворотки крови нормальный или умеренно повышен (в 3-5 раз от нормального), и при ЭМГ обычно выявляются признаки легкой миопатии. При гистологическом исследовании биоптата мышечной ткани выявляются хронические миопатические изменения, но может присутствовать мононуклеарная воспалительная инфильтрация (Rothstein et al., 1971).
Более чем у 95% пациентов с лице-плече-лопаточной дистрофией (ЛПЛД) имеется делеция в повторяющемся регионе D4Z4 хромосомы 4q35 (Wijmenga et al., 1992), что выявляется при сравнении размеров фрагментов ДНК, полученных после ферментации с рестриктазой EcoR1. Молекулярные тесты широко применяются в молекулярных диагностических лабораториях. Фрагменты >42 килобаз считаются нормальными. Около 95% аномальных аллелей имеют 34 kb или меньше (Upadhyaya et al, 1997). Фрагменты в 38-41 kb считаются пограничными и оцениваются с учетом клинической картины.
Пациенты с меньшими фрагментами EcoRl (например, делеции больших объемов) имеют тенденцию к более раннему развитию и быстрому прогрессированию заболевания (Zatz et al., 1995). Примерно у 10-30% пробандов с лице-плече-лопаточной дистрофией (ЛПЛД) заболевание развивается в результате делеций, возникших de novo; у остальной части пациентов имеется семейный анамнез, соответствующий аутосомно-доминантному механизму наследования. В немногих семьях заболевание не связано с 4q35, что свидетельствует о его генетической гетерогенности (Gilbert et al., 1993).
Многие расстройства могут проявляться мышечной слабостью, характер которой напоминает лице-плече-лопаточная дистрофия (ЛПЛД). Миофибриллярная миопатия, миозит с включенными тельцами, митохондриальные миопатии и врожденные миопатии обычно дифференцируются на основании гистопатологического исследования мышечной ткани. Иногда сложно дифференцировать лице-плече-лопаточную дистрофию (ЛПЛД) от некоторых форм конечностно-поясничной мышечной дистрофии (особенно КПМД 2А) и полимиозита. Миотонические дистрофии могут манифестировать ЛПЛД-подобным синдромом, дифференцировка обычно проводится на основании данных ЭМГ.