Конечностно-поясничные мышечные дистрофии (КПМД) у ребенка
Конечностно-поясничные мышечные дистрофии (КПМД) — это гетерогенная группа заболеваний, характеризующаяся слабостью и истончением мышц плечевого пояса и пояса нижних конечностей и дистрофическими изменениями в биоптате, указывающими на продолжающуюся дегенерацию мышечных волокон, вариабельность размера мышечных волокон и увеличение объема соединительной ткани.
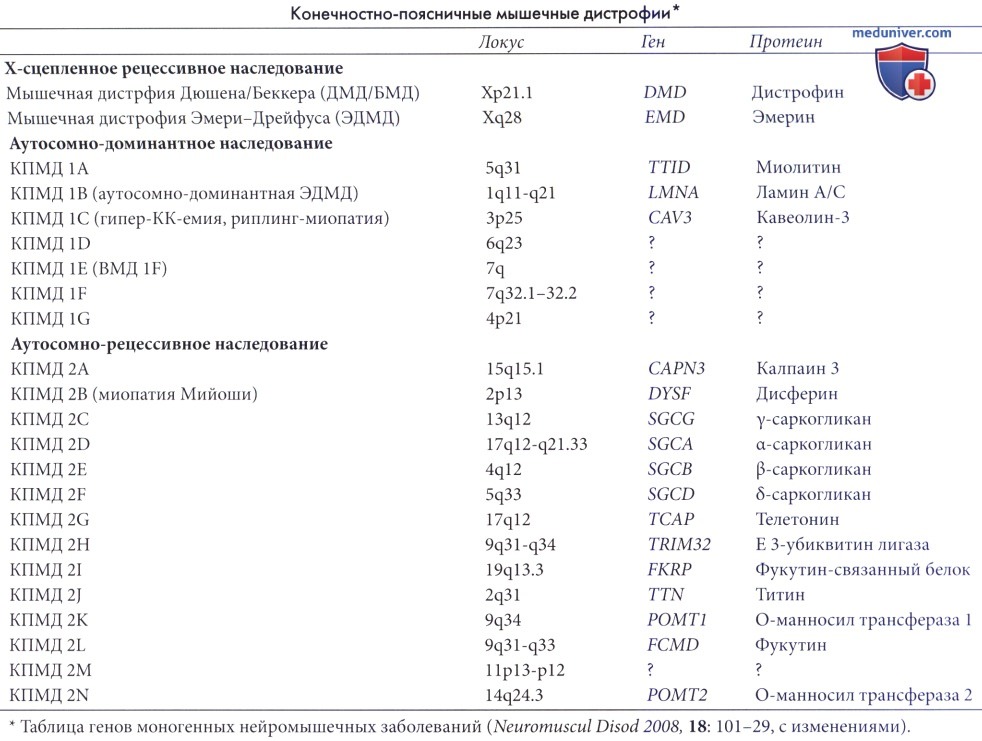
Даже среди пациентов с одним специфическим генотипом клиническое течение варьирует от тяжелых форм с быстрым прогрессированием и началом болезни в первой декаде (сходных по клиническим проявлениям с мышечной дистрофией Дюшена), до более легких форм, прогрессирующих медленнее и с более поздним началом (сходные по тяжести с мышечной дистрофией Беккера) (подробные обзоры см. Gordon и Hoffmann, 2001; Zatz et al., 2003; Laval и Bushby, 2004). В настоящее время идентифицированы 19 генных локусов, ответственных за развитие конечностно-поясничные мышечные дистрофии (КПМД).
Помимо этого существуют другие формы мышечной дистрофии с поздним началом, которые необходимо учитывать при дифференциальной диагностике в клинике.
а) Корреляция клинической картины и принципы диагностики. Детальный разбор всех разнообразных форм конечностно-поясничной мышечной дистрофии (КПМД) не входит в задачи этого руководства. Тем не менее, существуют несколько простых клинических признаков, которые помогут врачу при диагностике и укажут направление дальнейших лабораторных исследований. В целом типична межсемейная и внутрисемейная вариабельность. Гипертрофия икроножных мышц может наблюдаться во всех группах, но при конечностно-поясничной мышечной дистрофии (КПМД) 2В она встречается редко, при этой форме болезни может развиться атрофия икр.
Поражение сердца (кардиомиопатия) часто развивается при ДМД, ЭДМД, КПМД 1В, саркогликанопатиях и КПМД 21, но редка при КПМД 2А и 2В. Тяжелый (дюшеноподобный) фенотип может наблюдаться при саркогликанопатиях и КПМД 2А и 21.
Разные формы конечностно-поясничной мышечной дистрофии (КПМД) иногда невозможно различить на основании клинических проявлений. Диагноз зачастую устанавливается на основании исследований протеинов биоптата мышечной ткани пациента (иммуногистохимические исследования и вестерн-блоттинг) с целью выявления дефицита специфических протеинов; после этого выполняется анализ на наличие мутаций, подтверждающий, что дефицит протеинов вызван первичной мутацией в соответствующем гене. Следует отметить, что анализ протеинов не всегда специфичен — например, патология дисферина выявляется почти у 40% больных конечностно-поясничной мышечной дистрофией (КПМД), хотя полное его отсутствие, выявляемое при иммуногистохимическом исследовании (ИГХ), обычно вызывается первичной мутацией дисферина (Bansal и Campbell, 2004).
Первичные мутации в каком-либо из комплексов саркогликанов, могут привести к дефициту всех компонентов саркогликанового комплекса, выявляемой при ИГХ.
Специфичность дефицита калпаина, выявляемого при вестерн-блоттинге, составляет 70%, но также может возникать и при других дистрофиях; и наоборот, пациенты с мутациями в гене калпаина были выявлены среди тех, у кого реакция вестерн-блоттинга на калпаин была нормальной. Лаборатории, осуществляющие исследование ДНК с целью диагностики (т.е. платно) или с исследовательской целью, перечислены на www.geneclinics.org.
б) Патогенез. Как уже говорилось выше, мышечная дистрофия Дюшена вызывается дефицитом мембранного протеина сарколеммы — дистрофина. После открытия дистрофина исследователи обратили внимание на протеины, связанные или взаимодействующие с дистрофином, так как была высока вероятность того, что эти протеины тоже участвуют в патогенезе нервно-мышечных расстройств человека. Это привело к клонированию генов дистрофинсвязанных протеинов (ДСП), α-, β-, δ- и γ-саркогликанов и идентификации мутаций в каждом гене при различных формах АР-КПМД (КПМД 2C-F). Была выдвинута гипотеза, что недостаточность какого-либо компонента комплекса ДСП нарушает связывание экстрацеллюлярного матрикса и цитоскелета, что приводит к нестабильности сарколеммы и, как результат, к некрозу (Zatz et al., 2003; Laval и Bushby, 2004).
Конечностно-поясничная мышечная дистрофия (КПМД) 2I связана с потерей α-дистрогликана клеточной мембраны, что также приводит к нарушению связи между матриксом и цитоскелетом. Гликозилирование α-дистрогликана различными гликозилтрансферазами обеспечивает его нормальную локализацию в мембране мышечного волокна (Muntoni et al., 2002). КПМД 21 вызывается рецессивной мутацией в гене, кодирующем гликозилтрансферазу ФСП (фукутин-связанного протеина). Аберрантное гликозилирование α-дистрогликана также лежит в основе патогенеза гетерогенной группы врожденных мышечных дистрофий с поражением центральной нервной системы и без такового (например, ВМД Фукаямы, синдрома Уокера-Варбурга, болезни мышц-глаз-мозга, ВМД тип 1C).
Мутации в гене, кодирующем протеин дисферин, вызывают КПМД 2В и миопатию Мийоши (первоначально поражающую преимущественно мышцы дистальныхэкстрацеллюлярным отделов). Дисферин локализуется на сарколемме, но не является неотъемлемым компонентом комплекса ДСП. Он участвует в процессах слияния везикул в скелетных мышцах и, как было недавно установлено, в процессах репарации мышечной мембраны (Bansal и Campbell, 2004). Это указывает на другой механизм поражения мышцы, т.е. скорее неадекватное восстановление, а не повышенную восприимчивость к повреждению мембраны (как это наблюдается при дистрофиях, вызванных патологией дистрофина и комплекса ДСП).
Помимо этого было выявлено несколько генов мышечных дистрофий, не кодирующих неотъемлемые компоненты комплекса ДСП, а именно калпаин, калций-чувствительная внутриклеточная протеаза; кавеолин 3, компонент мембранных липидных рафтовых доменов; ламин А/С и эмерин, протеины ядерной мембраны; протеины саркомеров телетонин и миотилин; TRIM3, пируват убиквитин лигаза (Zatz et al., 2003). Патология этих протеинов может вызвать разрыв цепей клеточного метаболизма, и именно это будет основным звеном патогенеза, а не вызванные этими изменениями дистрофические процессы мышечной ткани вследствие прямого нарушения структуры и целостности сарколеммы.
в) Врожденная мышечная дистрофия (ВМД). Термином врожденная мышечная дистрофия (ВМД) обозначают гетерогенную группу наследственных расстройств, характеризующихся мышечной слабостью с рождения или в первые месяцы жизни, обычно с картиной дистрофии при гистологическом исследовании биоптата мышечной ткани и повышенным уровнем креатинкиназы (хотя иногда гистологически может выявляться миопатия, а не дистрофия, и уровень КК может быть в пределах нормы). У младенцев часто наблюдаются гипотония и контрактуры суставов. Обычно заболевание протекает статично, хотя могут отмечаться улучшение или медленное прогрессирование.
Поражения дыхательной мускулатуры, сердечной мышцы и мышц глаза вариабельны (Dubowitz, 1995; Jones и North, 2003). Современное рабочее определение ВМД, предложенное Voit, представлено в таблице ниже.
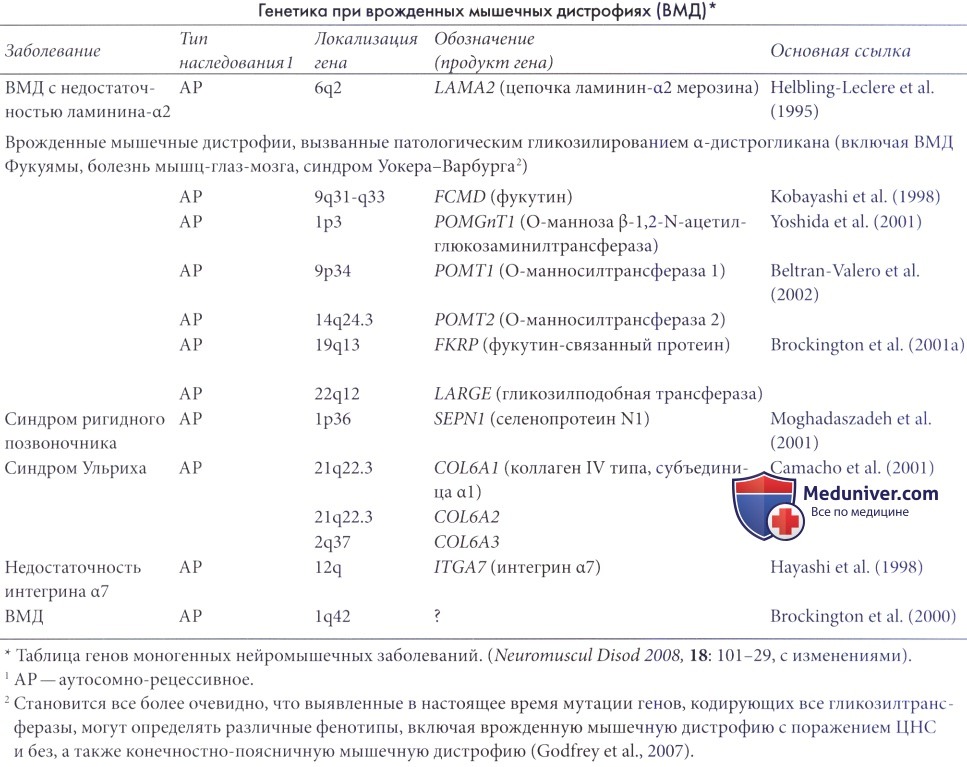
Известные на данный момент гены, вызывающие ВМД, представлены в таблице ниже. В настоящее время идентифицированы десять генов, вызывающих специфические формы ВМД. Необходимо отметить, что многие случаи ВМД остаются неклассифицированными.
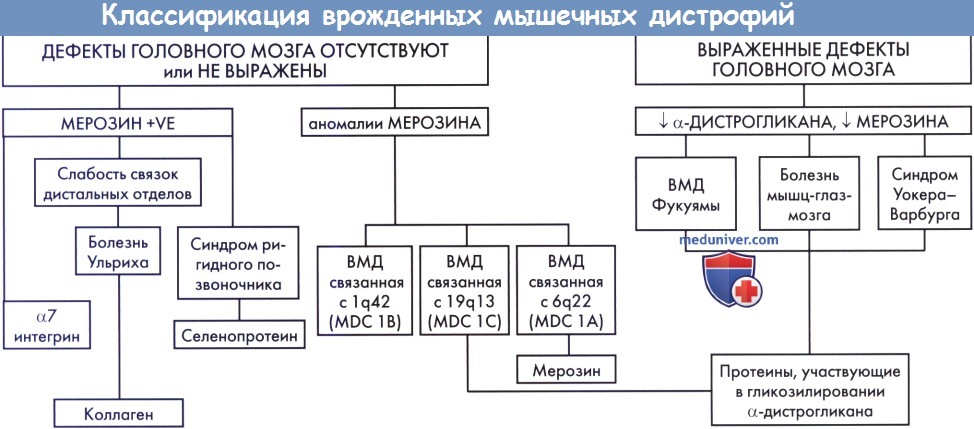
Первоначально классификация ВМД основывалась на клинических проявлениях. Однако в настоящее время классификация базируется на комбинации клинических, биохимических и молекулярных генетических признаках, поскольку многие формы ВМД проявляются одинаково (перекрывающие фенотипы). Врожденные мышечные дистрофии можно разделить на две большие группы: 1) без патологии головного мозга (без поражения ЦНС) и 2) с выраженной патологией головного мозга. Эти группы могут далее подразделяться на основании экспрессии экстрацеллюлярных матричных протеинов, ламинина-α2, известного также как мерозин, а также на основании клинических признаков, таких как слабость связок дистальных отделов конечностей (часто встречающаяся при патологии коллагена IV) и ригидный позвоночник (при расстройствах селенопротеина N).
г) Врожденные мышечные дистрофии без выраженной патологии головного мозга: α-декстрогликанопатии:
1. Первичная недостаточность ламинина-α2 (мерозина) (MDC1A). У детей с первичной недостаточностью ламинина-α2 заболевание обычно проявляется при рождении или вскоре после него сильно выраженной общей гипотонией, нарушениями дыхания и трудностями при кормлении. Тяжелая мышечная слабость поражает мышцы лица, туловища и конечностей, и примерно у половины пациентов имеются контрактуры одного или нескольких суставов. Мышечная слабость не склонна к прогрессированию, или прогрессирует медленно, однако некоторые пациенты способны стоять самостоятельно (Voit, 2001; Jones и North, 2003).
У всех пациентов с тяжелыми аномалиями ламина-α2 отмечаются изменения белого вещества головного мозга или изменения при МРТ, напоминающие лейкодистрофию, хотя интеллект обычно остается нормальным или отмечается легкая задержка интеллектуального развития. Изменения белого вещества становятся более выраженными в первые годы жизни, параллельно с усиленной миелинизацией головного мозга, но затем остаются неизменными. У некоторых пациентов присутствуют дополнительные структурные аномалии, включая затылочную полимикрогирию/агирию и мозжечковую или мостовую гипоплазию. Почти у 30% развивается эпилепсия. Уровни сывороточной КК обычно повышены, от 1000 до 15000 МЕ/л. Описаны случаи с поздним началом и легкой мышечной слабостью (Jones et al., 2001; Miyagoe-Suzuki et al., 2000). При гистологическом исследовании биоптата мышечной ткани выявляются тяжелые дистрофические изменения, при иммуногистохимическом исследовании количество ламина-α2 значительно снижено или он отсутствует вообще.
Заболевание наследуется по аутосомно-рецессивному механизму и вызывается мутацией гена LAMA2 хромосомы 6q (Helbling Leclerc et al., 1995). Необходимо отметить, что при иммуногистохимическом исследовании и при количественной оценке с помощью вестерн-блоттинга могут выявляться вторичные патологические изменения, возникшие вследствие нарушений гликозилирования α-дистрогликана. В последнем случае окрашивание ламина-а2 обычно неоднородно и ослаблено, а не отсутствует полностью.
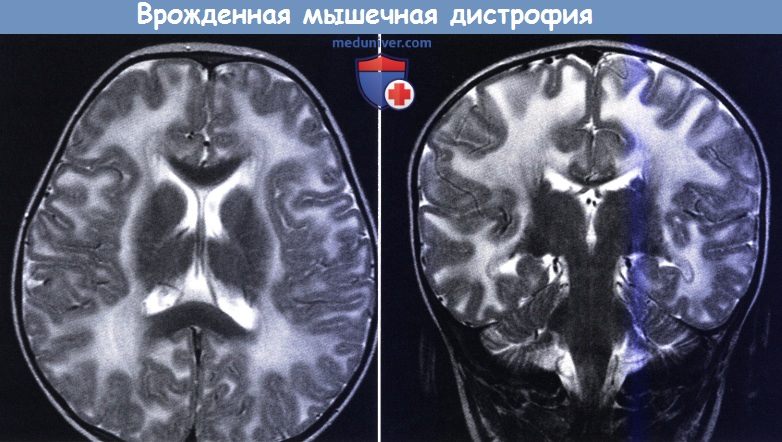
Врожденная мышечная дистрофия с недостаточностью мерозина.
На МРТ виден диффузно усиленный Т2 сигнал от белого вещества. Аксиальный (слева) и фронтальный срез (справа).
2. Синдром ригидного позвоночника. Это расстройство аллельно болезни множества мелких стержней; клиническая картина одинакова у пациентов с мутациями гена, кодирующего селенопротеин N (SEPN1), с мышечной слабостью преимущественно осевой мускулатуры и выраженной ригидностью позвоночника. Подобная ригидность не специфична для этого заболевания, но наблюдается и при других фенотипах, например при мышечной дистрофии Эмери-Дрейфуса. Распространенность мышечной слабости, выраженность сколиоза и контрактур суставов и тяжесть функциональных нарушений вариабельны. Дыхательная недостаточность, требующая проведения ночной искусственной вентиляции, встречается часто и может наблюдаться у самостоятельно обслуживающих себя пациентов, поэтому большое значение имеет ночной мониторинг гиперкапнии. Интеллект и результаты лучевых исследований головного мозга обычно нормальны, КК в пределах нормы (Voit 2001; Jones и North 2003).
3. Недостаточность интегрина α7. Это редкая форма ВМД и до настоящего момента первичные мутации гена интегрина α7 (ITGA7) были описаны только у трех пациентов (Hayashi et al., 1998).
4. Врожденная мышечная дистрофия Ульриха (УВМД). Впервые это заболевание было описано Ульрихом в 1930 г. как врожденная гипотоническая-склеротическая мышечная дистрофия. Основные клинические проявления включают генерализованную мышечную слабость, истощение и гипотонию, ярко выраженные контрактуры в проксимальных суставах и чрезмерную растяжимость в дистальных отделах с раннего младенческого возраста, прогрессирующее течение с медленным нарастанием слабости и усилением контрактур (Higuchi et al., 2001). Другие клинические признаки болезни включают в себя сухость кожи и сыпь «по типу наждачной бумаги», которую часто ошибочно принимают за экзему (Bertini и Рере, 2002), высокое небо и проминенцию пяток. Интеллектуальное развитие обычно нормально. Также могут наблюдаться двусторонние врожденные вывихи бедер, кривошея и кифосколиоз (Camacho Vanegas et al., 2001; Higuchi et al., 2001; Bertini и Pepe, 2002; Pepe et al., 2002).
Часто они сопровождаются рано развивающимися ригидностью позвоночника и нарушениями дыхания. Некоторые пациенты с возрастом обретают способность к самообслуживанию, другие — нет (Mercury et al., 2002). УВМД вызывается мутацией в одном из трех генов, кодирующих компоненты протеина коллагена IV (COL6A1, 2 и 3). При исследовании биопсии выявляются типичные патологические изменения мышечной дистрофии (Higuchi et al., 2001), количество коллагена IV в сарколемме в различной степени снижено. Уровень сывороточной КК нормальный или слегка повышен.
Миопатия Бетлема (БМ) (Higushi et al., 2001) — более легкая патология коллагена IV, с поздним началом и менее выраженной мышечной слабостью в сочетании с избыточной подвижностью связочного аппарата дистальных отделов и контрактуры. Изначально УВМД считалась аутосомно-рецессивным заболеванием, а миопатия Бетлема — более легким аутосомно-доминантным вариантом. Однако в ходе недавних генетических исследований было выявлено, что почти в 40% случаев УВМД может вызываться доминантными мутациями, и что эти мутации часто возникают de novo. Также оказалось, что степень тяжести клинической картины болезней коллагена IV постоянна, поэтому не всегда возможно четко отдифференцировать УВМД от БМ, и в некоторых случаях врожденной мышечной слабости клиническое течение болезни бывает относительно легким (Baker et al., 2005; Lampe et al., 2005).
5. Врожденные мышечные дистрофии с выраженными аномалиями головного мозга: α-дистрогликанопатии. Некоторые ВМД вызываются мутациями в генах, кодирующих гликозилтрансферазы или пируват-гликозилтрансферазы. Аномальное гликозилирование α-дистрогликана — часто встречающееся нарушение, приводящее к аномальной локализации α-дистрогликана в мышечной мембране. Это проявляется в ослаблении или отсутствии прокрашивания α-дистрогликанов при иммуногистохимическом исследовании. Ламинин-α2 связывается с α-дистрогликаном на клеточной мембране, что вызывает вторичное снижение количества ламинина-2; иммуногистохимическое прокрашивание может быть неравномерным или нормальным, но по результатам вестерн-блоттинга содержание этого протеина может быть значительно снижено.
При ВМД, вызванных аномальным гликозилированием α-дистрогликана, развиваются различные поражения центральной нервной системы (обычно — нарушение миграции нервных клеток) и глазные симптомы; эти болезни включают в себя ВМД Фукуямы, болезнь мышц-глаз-мозга, синдром Уокера-Варбурга, MDC1C и MDC1D. Клинические картины этих различных состояний в значительной степени перекрывают друг друга, также выявлена генетическая гетерогенность (Jones и North, 2003; Godfrey et al., 2007).
6. Врожденная мышечная дистрофия Фукуямы (ФВМД). ФВМД встречается преимущественно в Японии. Болезнь обычно манифестирует во внутриутробном или в неонатальном периоде снижением подвижности и тяжелой дистрофической патологией. Некоторым пациентам удается научиться ходить самостоятельно. Примерно у половины выявляются заболевания глаз. Часто встречающиеся аномалии миграции нервных клеток включают полимикрогирию, пахигирию и агирию большого мозга и мозжечка. У большинства наблюдаются нарушения интеллекта и припадки. У большинства пациентов развиваются поражения сердца и нарушения дыхания, из-за чего сильно сокращается ожидаемая продолжительность жизни (Jones и North, 2003; Muntoni и Voit, 2004). ФВМД наследуется по аутосомно-рецессивному механизму и вызывается мутацией в гене, локализованном в хромосоме 9q31, кодирующем фукутин, мнимую гликозилтрансферазу (Schachter et al., 2003; Muntoni et al., 2004).
7. Болезнь мышц-глаз-мозга (МГМ). МГМ по тяжести сходна с ФВМД. Клинические проявления включают морфологические дефекты глаз (микрофтальмия, дефекты сетчатки и аномалии передней камеры), лиссэнцефалию II типа (пахигирию, полимикрогирию, гипоплазию червя и кисты мозжечка) и ВМД. Часто наблюдаются припадки, нарушения интеллекта обычно тяжелые (Jones и North, 2003; Barresi et al., 2004; Muntoni и Voit, 2004). МГМ наследуется по аутосомно-рецессивному механизму (Muntoni и Voit, 2004). Вызывающие болезнь мутации впервые были выявлены в гене, кодирующем протеин гликозилтрансферазы О-манноз β-1,2-N-ацетилглюкозаминилтрансферазу (POMGnTl) хромосомы lq33 (Kano et al., 2002). Недавно была описана мутация гена POMGnT1 у пациента с аутистическими чертами и стереотипическими движениями, что указывает на еще более широкий спектр (Haliloglu et al., 2004).
МГМ — генетически гетерогенное заболевание; мутации, вызывающие МГМ, также были выявлены в гене FRKP (см. ниже), и вероятно, мутации генов, кодирующих другие гликозилтрансферазы определяют такой же фенотип (Beltran-Valero et al., 2004).
8. Синдром Уокера-Варбурга (УВС). УВС — наиболее тяжелая из мышечных дистрофий, характеризующихся патологией α-дитрогликана. У пациентов с УВС наблюдается ВМД, тяжелая патология глаз (микрофтальмия, гипоплазия зрительного нерва, дисплазия сетчатки и катаракты), лиссэнцефалия II типа с агирией, гипоплазией мозжечка и ствола головного мозга; обычно такие больные умирают в младенческом возрасте (Dubowitz, 1995; Jones и North, 2003; Currier et al., 2005). Другие сопутствующие врожденные мальформации включают гидроцефалию, мальформацию Дэнди-Уокера, расщепление губы или неба и аномалии гениталий (Dubowitz, 1995). УВС наследуется по аутосомно-рецессивному механизму и является генетически гетерогенным заболеванием (Muntoni и Voit, 2004; Beltran-Valero et al., 2004; Cohn, 2005; van Reeuwijk et al., 2005; Godfrey et al., 2007).
9. MDC1D. В литературе описан один пациент с ВМД, тяжелой задержкой умственного развития, патологическим гликозилированием α-дистрогликана и мутациями в гене LARGE, кодирующем еще одну гликозилтрансферазу. Уровень сывороточной КК был повышен. При МРТ головного мозга в возрасте 14 лет выявлены обширные и симметричные изменения белого вещества в перивентрикулярной области, распространяющиеся до дугообразных волокон. Также были выявлены изменения, указывающие на патологическую миграцию нервных клеток (Longman et al., 2003).
10. MDC1C. MDC1C вызывается мутацией гена FKRP (Brockington et al., 2001a). В первом описании у пациента наблюдалась тяжелая ВМД, проявлявшаяся в неонатальном периоде гипотонией, трудностями кормления и тяжелой мышечной слабостью. У некоторых развивается гипертрофия мышц (икроножных и языка), контрактуры и слабость мышц лица. У некоторых детей были тяжелые рестриктивные нарушения дыхания и нарушения функции сердца. Структура и функция головного мозга оставались нормальными. В большинстве случаев уровень КК был повышен, отмечалась дистрофия мышечной ткани.
Впоследствии были описаны дети с мутациями в FKRP, патологией структур головного мозга и нарушением его функции (de Paula et al., 2003; Topaloglu et al., 2003; Beltran-Valero et al., 2004; Harel et al., 2004; Louhichi et al., 2004), и оказалось, что такой фенотип встречается так же часто, как и первоначально описанный фенотип MDC1C. У пациента могут наблюдаться умственная отсталость и различные аномалии ЦНС, включая кисты мозжечка или его атрофию с поражением белого вещества или без него (Topalogu et al., 2003; Louhichi et al., 2004). Мутации FKRP также были описаны у одного пациента с болезнью МГМ и у одного пациента с УВС (Beltran-Valero et al., 2004).
Мутации FKRP также вызывают одну из наиболее часто встречающихся в Европе форм конечностно-поясничной мышечной дистрофии (КПМД 2I); у пациентов с КПМД 2I может развиваться дюшеноподобный фенотип или болезнь может позже дебютировать и проявляться более легким фенотипом с различной степенью тяжести, как это наблюдается и при мышечной дистрофии Беккера. При КПМД 2I часто наблюдаются дыхательные и сердечные нарушения (Brockington et al., 2001b).
Совокупность этих данных демонстрирует огромное разнообразие фенотипов, вызываемых мутациями FKRP, от случаев сохранного интеллекта до тяжелых мальформаций ЦНС и умственной отсталости, от больных с тяжелой ВМД, не способных научиться ходить самостоятельно, до случаев с бессимптомным течением вплоть до 30 лет и сохранением способности к самообслуживанию. Предполагается, что это зависит от степени нарушения функции протеина, вызванного мутацией (Beltran-Valero et al., 2004).