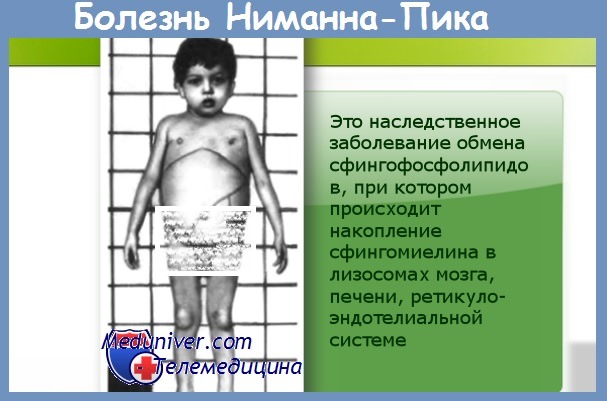
Болезнь Ниманна-Пика представляется собой гетерогенную группу заболеваний, характеризующихся накоплением сфингомиелина в ретикулоэндотелиальной системе. Из трех основных типов заболевания (А, В и С) А и В типы связаны с дефицитом кислой сфингомиелиназы и относятся к I типу, в то время как другие формы заболевания без дефицита сфингомиелиназы относятся ко II типу (классификация Schuchman и Desnick, 1995). Тип С будет рассмотрен вместе с нарушениями метаболизма холестерина.
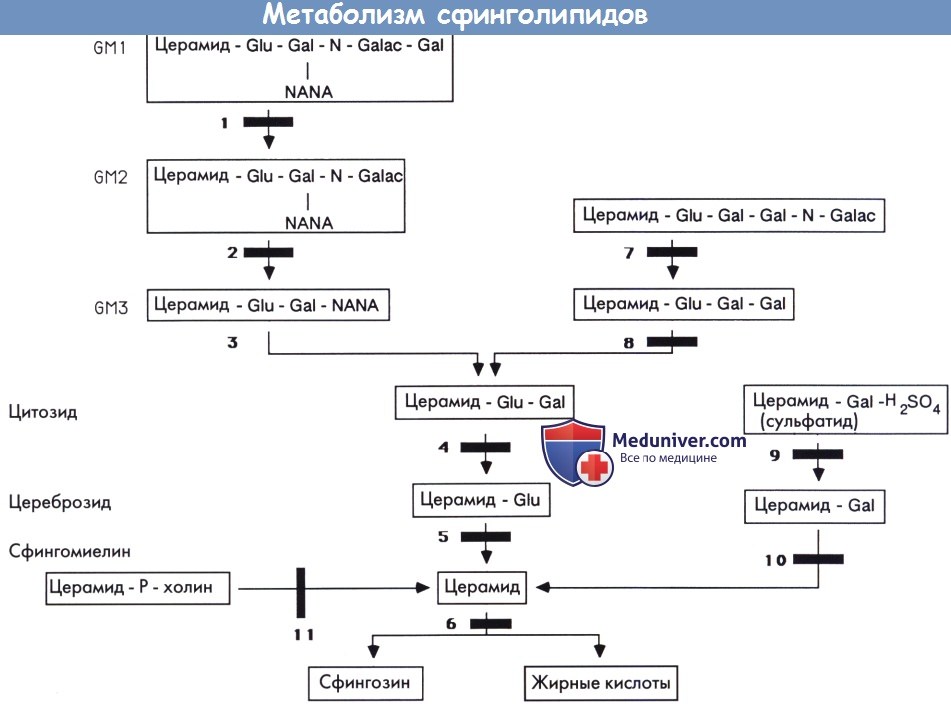
Метаболизм сфинголипидов.
Gal—галактоза. Glu—глюкоза. N-Galac—N-ацетилгалактозамин. NANA—нейраминовая кислота (N-ацетилнейраминовая кислота).
1 — β-галактозидаза. 2 — β-гексозаминидаза А (болезнь Тея-Сакса) и другие ганглиозидозы типа II GМ2.
3 — GM3 сиалидаза. 4 — лактозилцекамидаза (лактозилцерамидоз). 5 — β-глюкозидаза (глюкоцереброзидаза) (болезнь Гоше).
6 — церамидаза (болезнь Фарбера). 7 — β-гексозаминидаза В (болезнь Сандгоффа и другие О варианты GM2 ганглиозидозов).
8 — церамид-тригексозидаза/α-галактозидаза (болезнь Фабри). 9 — цереброзидсульфатаза/арилсульфатаза* (метахроматическая лейкодистрофия).
10 — β-галактозидаза/β-галактоцереброзидаза (болезнь Краббе). 11 — сфингомиелиназа (болезнь Ниманна-Пика А и В).
*Активность, оцененная на искусственных субстратах, не всегда соответствовала активности цереброзиодсульфатазы.
а) Болезнь Ниманна-Пика А типа (нейровисцеральный тип, тип I) наследуется аутосомно-рецессивным путем и встречается только у евреев ашкенази. Гены располагаются на участке 11 р 15; известно множество мутаций. Отмечена некоторая корреляция между определенной мутацией и фенотипом. Три мутации охватывают 92% случаев заболевания среди евреев ашкенази (Schuchman, 1995). Патологическим признаком заболевания является наличие в ретикулоэндотелиальной системе больших вакуолизированных клеток.
В центральной нервной системе обнаруживаются пенистые клетки и раздутые ганглионарные клетки. При биохимическом исследовании отмечается заметное накопление сфингомиелина, значимого компонента нормального миелина, в сочетании с накоплением холестерина в селезенке, печени и почках. Накопление, хотя и в умеренной степени, также отмечается в головном мозге.
Клинические признаки заболевания появляются в течение первого года жизни в виде гепатоспленомегалии и плохого физического и когнитивного развития. Часто встречаются желтуха, диарея и инфильтраты в легких. Неврологические проявления выражены приблизительно в трети случаев и чаще всего появляются со временем. Основными признаками являются миоклонические припадки, спастичность и слепота.
У четверти пациентов выявляется симптом вишневой косточки. Периферическая нейропатия с замедлением проводимости отмечается в 10% случаев. Смерть обычно наступает в возрасте до 5 лет.
Диагноз можно предположить на основании наличия вакуолизированных клеток в костном мозге. Дефицит сфингомиелиназы в лейкоцитах или фибробластах подтверждает диагноз.
б) Болезнь Ниманна-Пика В типа (висцеральный тип) характеризуется поражением висцеральных органов без неврологических проявлений и встречается у детей старшего возраста и взрослых. У отдельных пациентов могут развиться неврологические признаки.
В действительности четкое разделение типов А и В не всегда возможно. Известны вялотекущие нейровисцеральные формы с многолетним течением; в ходе недавних крупных исследований чаще всего выявлялись промежуточные формы, которые были зарегистрированы у 12 из 25 пациентов (Pavlu-Pereira et al., 2005). Проводятся исследования терапии миглустатом.
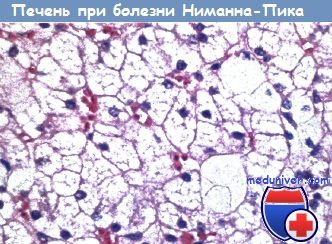
Болезнь Ниманна-Пика (сфингомиелиново-холестериновый липидоз). Микрофотография при большом увеличении.
Слои увеличенных пенистых клеток Купфера, заполненных сфингомиелином-холестерином.