Функция лизосом заключается в гидролизе ряда сложных молекул. Нарушение данной функции приводит к нарушению накопления внутри лизосом. Большая часть лизосомальных болезней связана с генетическим дефектом одного из лизосомальных ферментов, участвующих в расщеплении специфического вещества. Тем не менее, некоторые заболевания, включая муколипидоз I и II, являются результатом посттрансдукционных аномалий, затрагивающих нормально синтезированные проферменты. Другие заболевания (например, болезнь накопления сиаловых кислот или цистиноз) связаны с дефектом транспорта субстратов через мембрану лизосом.
Все лизосомальные болезни наследуются рецессивным путем (преимущественно аутосомно-рецессивным). Новые заболевания (например, нейрональный восковидный липофусциноз) все еще добавляются к данному списку.
Клинические проявления варьируют в зависимости от дефектного фермента. Даже при дефиците одного и того же фермента фенотипические проявления могут быть различными, что отражает возможность наличия множества различных аномалий того же гена и/ или совместное действие других генов, альтернативного сплайсинга транспортной РНК или посттрансдукционных модификаций, что в свою очередь приводит к отсутствию или различным аномалиям белков, различной их активности и, как следствие, различным клиническим проявлениям.
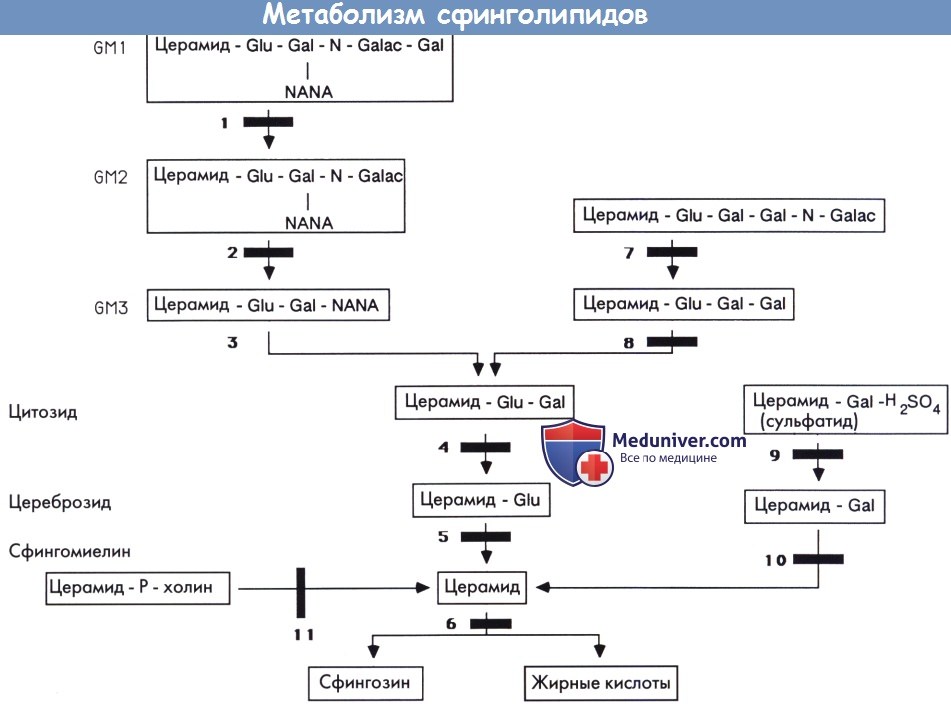
Сфинголипидозы. Сфинголипидоз является лизосомальной болезнью, проявляющейся отсутствием или неполным распадом сфинголипидов, которые являются необходимыми компонентами мембран центральной нервной системы. Основные этапы катаболизма сфинголипидов, соответствующие ферменты и ферментные блоки представлены на рисунке ниже.
Ганглиозидозы. Имеется ряд важных заболеваний, развивающихся в результате ферментного блока, затрагивающего удаление N-ацетилгалактозы из сложных молекул ганглиозидов. Гинглиозиды находятся преимущественно в области ядер серого вещества и не обнаруживаются (в сколько-нибудь значительном количестве) в миелине. Функция данных веществ в норме не до конца понятна. Несмотря на то, что из головного мозга выделены многие ганглиозиды, четыре основных компонента (от GM1 до GM4) составляют более 90% общей фракции ганглиозидов. Возлагается надежда на эффективную терапию при применении имино-сахарных ингибиторов гликозил-церамидтрансферазы, которые эффективны при болезни Гоше, и, возможно, будут эффективны при других нарушениях обмена гликосфинголипидов (Aerts et al., 2006).
Метаболизм сфинголипидов.
Gal—галактоза. Glu—глюкоза. N-Galac—N-ацетилгалактозамин. NANA—нейраминовая кислота (N-ацетилнейраминовая кислота).
1 — β-галактозидаза. 2 — β-гексозаминидаза А (болезнь Тея-Сакса) и другие ганглиозидозы типа II GМ2.
3 — GM3 сиалидаза. 4 — лактозилцекамидаза (лактозилцерамидоз). 5 — β-глюкозидаза (глюкоцереброзидаза) (болезнь Гоше).
6 — церамидаза (болезнь Фарбера). 7 — β-гексозаминидаза В (болезнь Сандгоффа и другие О варианты GM2 ганглиозидозов).
8 — церамид-тригексозидаза/α-галактозидаза (болезнь Фабри). 9 — цереброзидсульфатаза/арилсульфатаза* (метахроматическая лейкодистрофия).
10 — β-галактозидаза/β-галактоцереброзидаза (болезнь Краббе). 11 — сфингомиелиназа (болезнь Ниманна-Пика А и В).
*Активность, оцененная на искусственных субстратах, не всегда соответствовала активности цереброзиодсульфатазы.
а) Ганглиозидозы типа II GМ2 являются наиболее распространенными заболеваниями в данной группе. Известно, что несколько вариантов заболевания зависят от природы дефекта фермента. Выявлено три изофермента гексозаминидазы. Гексозаминидаза А состоит из двух альфа и двух бета субъединиц (A1, В2), гексозаминидаза В состоит только из В субъединиц (В2, В2), а гексозаминидаза S—только из А субъединиц (А2). При классическом варианте болезни Тея-Сакса или В-варианте гексозаминидаза А и S становятся неэффективны в результате мутации альфа-локуса 15-йхромосомы (15q22-q25.1).
Гексозаминидаза В функционирует нормально, но не способна гидролизовать ганглиозиды в условиях in vivo. Известны различные мутации альфа-локуса. Таким образом, ганглиозидозы типа II GМ2 у франко-канадцев могут быть связаны и отличаются от дефекта ДНК (делеция интрона 1 и области промотора) у евреев ашкенази, который в 73% случаев представляет собой вставку четырех оснований в экзон 11. Тем не менее, у некоторых франко-канадцев выявляется та же мутация, что и у евреев ашкенази, также могут отмечаться разные мутации внутри одной популяции. Мутация В1 приводит к нарушению специфичности к субстрату гексозаминидазы А. В данном варианте мутантный фермент демонстрирует в целом нормальную активность при тестировании с общепринятым метилумбеллиферил субстратом, но не способен гидролизовать GM2 или синтетический субстрат сульфат умбеллиферила.
Болезнь Сандгоффа характеризуется дефицитом обоих изоферментов бета-гексозаминидазы А и В в связи с мутацией в бета-локусе хромосомы 5q13. Причиной варианта АВ является дефицит белка активатора (сапонина), необходимого для взаимодействия гексозаминидазы А с ее естественным субстратом (Cordeiro et al., 2000). Таким образом, отмечается значимая клиническая и биохимическая гетерогенность среди ганглиозидов GM2 (Lyon et al., 2006).
Патологические изменения центральной нервной системы типичны для всех форм ганглиозидозов; отмечаются некоторые варианты поражения, связанные с длительностью течения и неопределенными факторами. Основным изменением является присутствие в цитоплазме нейронов жирорастворимых веществ с дальнейшим исчезновением многих нейронов и формированием распространенного глиоза. Также поражаются клетки Пуркинье, нейроны ядер ствола мозга и нейроны висцерального сплетения. Признаки воспаления явно выражены и совпадают с наличием клинических симптомов, что предполагает роль воспаления в повреждении головного мозга (Jeyakumar et al., 2003). Поражение внутренних органов, в особенности почек, заметно при болезни Сандгоффа, проявлением которой является обширное скопление глобозидов.
При электронной микроскопии выявляются множественные мембранные цитоплазматические тельца (тельца Терри), состоящие преимущественно из липидов и 10% белка. Отростки пораженных нейронов распространены на большой площади, и избыток легко возбудимых мембран может быть причиной аномальной функции нейронов.
1. Болезнь Тея-Сакса является наиболее частой формой ганглиозидозов и встречается у 1 на 2000 человек среди евреев ашкенази в восточной Европе. Частота встречаемости гена составляет 1 на 27 среди евреев ашкенази и 1 на 380 среди неевреев.
Заболевание начинается в период с 3 до 9 месяцев в виде потери приобретенных навыков и мышечного тонуса после полностью нормального исходного развития. В течение нескольких месяцев заболеванию может предшествовать реакция вздрагивания. Неврологические симптомы быстро прогрессируют. Исходная гипотония сменяется спастическим тетрапарезом, на поздних стадиях заболевания могут появиться эпилептические припадки. После первого года жизни дети беспомощны, слепы, не реагируют на внешние раздражители, часто развивается прогрессирующая макроцефалия. На ранних стадиях с помощью МРТ можно выявить усиления сигнала базальных ганглиев в Т2-режиме. Хвостатое ядро часто выступает в боковые желудочки. Позднее сигнал в Т2-режиме усиливается в области белого вещества (Grosso et al., 2003).
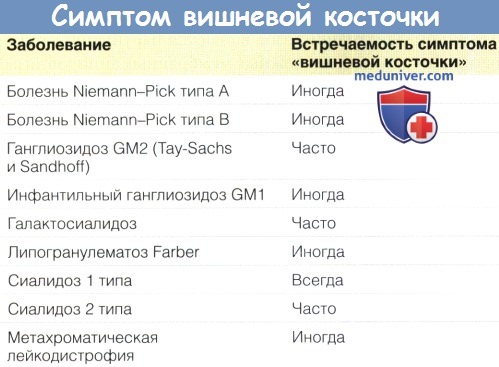
Смерть обычно наступает до трех лет. Уже на ранних стадиях заболевания отмечается симптом вишневой косточки в обеих макулах, окруженный зоной молочно-белой сетчатки. Формирование данного белого участка связано с накоплением липидов в ганглионарных клетках сетчатки, которые особенно плотно расположены в области заднего полюса, в то время как красное пятно соответствует нормальной макуле, в которой нет ганглионарных клеток, и представляет собой аномально красный, контрастирующий с окружающей сетчаткой участок. Несмотря на характерное проявление болезни Тея-Сакса, симптом вишневой косточки может отмечаться и при других типах ганглиозидозов, сиалидозе, болезни Ниманна-Пика, болезни Гоше младенцев и, в редких случаях, при метахроматической лейкодистрофии и болезни Краббе (Naidu et al., 1988).
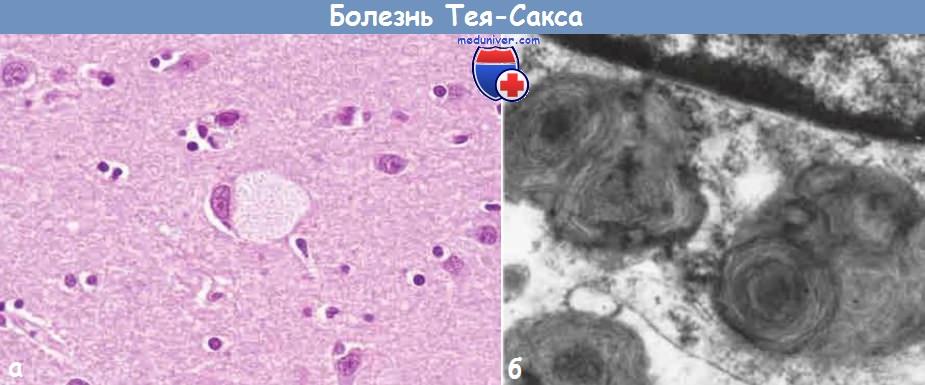
Ганглионарные клетки при болезни Тея-Сакса.
(А) Под световым микроскопом видно, что крупный нейрон содержит липидные вакуоли.
(Б) При электронной микроскопии в части нейрона видны лизосомы извитой формы. Сверху — часть ядра.
2. Болезнь Сандгоффа (ганглиозидоз 0 типа GM2) клинически не отличается от классической болезни Тея-Сакса, но составляет только 7% случаев GM2 ганглиозидозов.
Диагноз болезни Тея-Сакса легко подтверждается отсутствием гексозаминидазы А или гексозаминидаз А и В в сыворотке или лейкоцитах. Среди пациентов с В1 мутацией при оценке с несульфатированным умбилиферрильным субстратом отмечается промежуточный уровень активности гексозаминидазы А. В таких случаях необходимо использовать для диагностики 4-Mu-галактозо-N-ацетил-6-сульфат (4-MUG) (Matsuzawa et al., 2003). Во втором триместре беременности возможна пренатальная диагностика путем определения содержания гексозаминидазы А.
Возможно выявление носителей на основании анализа крови; данный метод использовался при автоматизированных скрининговых исследованиях (Kaback et al., 1993; Leib et al., 2005). Оценка фибробластов требуется в случае беременности или при наличии у партнера диабета, гепатита или перенесенного инфаркта миокарда. Псевдодефицит гексозаминидазы А встречается у сложных гетерозигот, которые являются носителями общей болезнетворной мутации в одном аллеле и двух доброкачественных мутаций во втором аллеле. Такие люди клинически здоровы, несмотря на неспособность гидролизовать синтетический субстрат 4-MUG. Данные доброкачественные мутации имеют значение для программ скрининга гетерозигот и пренатальной диагностики. Частота таких мутаций составляет 3% среди евреев ашкенази и 38% среди неевреев-носителей определенных ферментов (Cao et al., 1993).
3. Ювенильные ганглиозидозы типа II GМ2 встречаются редко. Заболевание дебютирует в возрасте 2-6 лет в виде нестабильности походки и часто в виде нарушений речи с последующим развитием атаксии и пирамидных знаков. В конечной стадии заболевания отмечается расстройство интеллекта (Hendriksz et al., 2004). Первыми проявлениями могут быть атаксия и дизартрия. Могут отмечаться припадки, зарегистрированы случаи дистонических и хореоатетоидных движений (Nardocci et al., 1992). Заболевание не связано с принадлежностью к какой-либо расе.
4. Ганглиозидозы типа II GМ2 с поздним началом (Neudorfer et al, 2005) также называются хроническими GM2 ганглиозидозами или «взрослой формой», несмотря на то, что в 35% случаев симптомы появляются до 10-летнего возраста. Заболевание начинается с особых форм дизартрии с дальнейшим формированием нарушений походки, пирамидных знаков, атаксии и поражения нижних мотонейронов. В половине случаев позже развиваются психиатрические нарушения. У некоторых пациентов отмечается дистония, зарегистрированы случаи супрануклеарной офтальмоплегии (Rucker et al., 2004).
Атипичные формы могут проявляться в виде болезни клеток передних рогов с началом в подростковом возрасте (Navon et al., 1995; Drory et al, 2003) в виде изолированной дистонии или атипичной спиноцеребеллярной дегенерации. Известны необычные проявления, имитирующие опухоль ствола мозга (Nassogne et al., 2003). Миоклонические припадки отмечаются в случае АВ варианта (Sakuraba et al., 1999).
Клиническая картина болезни Тея-Сакса может отмечаться у пациентов с нормальным количеством гексозаминидаз А и В (АВ вариант) (Cordeiro et al., 2000; Lyon et al., 2006) и плохо поддается диагностике, так как требует непосредственного исследования катаболизма меченых натуральных ганглиозидов. В1 вариант обычно проявляется в детском возрасте, но также может обнаруживаться у пациентов с подростковыми и хроническими формами (Grosso et al., 2003), тем не менее, В1 и АВ варианты могут встречаться не так редко.
В настоящее время не существует эффективного лечения GM2 ганглиозидозов. В ходе исследований на животных была продемонстрирована частичная эффективность терапии, направленной на уменьшение количества субстрата в сочетании с ингибитором синтеза церамидов миглустатом (Andersson et al., 2004; Сох, 2005). Процесс накопления ганглиозидов и последующая дегенерация головного мозга уже достаточно выражены во втором триместре внутриутробного развития, то есть терапия замещения ферментов не является предпочтительной.
б) GM1 ганглиозидозы. Данная группа заболеваний вызвана дефицитом кислой бета-галактозидазы, ген которой расположен на участке 3pter-3p21. Кроме накопления липидов, сходного с проявлениями GM2 ганглиозидозов, накапливаемый материал содержит также вакуолярные включения с аккумуляцией ряда содержащих маннозу олигосахаридов, что является результатом дефекта удаления терминальных бета-галактозных остатков во время катаболизма гликопротеидов. Известно несколько изоферментов бета-галактозидазы. При всех формах GM1 галактозидозов отмечается дефект трех изоферментов (A1, А2, А3), а различные фенотипы, вероятно, объясняются различными мутациями, приводящими к образованию белков с различной остаточной субстратной специфичностью (Suzuki et al., 1991; Lyon et al., 2006), тем не менее, корреляция между мутацией, остаточной активностью белка и клинической выраженностью симптомов отмечается не всегда.
В головном мозге выявляются патологические изменения в виде воспаления, как и при ганглиозидозах GM2, вероятно, играя роль в формировании клинических проявлений (Jeyakumar et al., 2003).
1. GM1 ганглиозидозы 1 типа (псевдоболезнь Гурлер, болезнь Лэндинга) являются редкими аутосомно-рецессивными заболеваниями, клинические признаки которых проявляются при рождении или даже пренатально (Tasso et al., 1996). У больных детей отмечается выраженная гипотония, плохое сосание, плохая прибавка в весе и отсутствие психомоторного развития, выступающие лобные бугры, грубые черты лица, макроглоссия и гирсутизм. У половины детей выявляется симптом вишневой косточки, обычно отмечается гепатоспленомегалия. Скелетные аномалии сходны с проявлениями синдрома Гурлера. Течение заболевания тяжелое. Возникают слепота, квадриплегия и эпилептические припадки, смерть обычно наступает до двухлетнего возраста.
2. GM1 ганглиозидозы 2 типа характеризуются поздним началом в возрасте от 6 месяцев до 3 лет. Значимых скелетных аномалий не отмечается, тем не менее, первый или второй шейный позвонок имеет аномальную форму, таким образом, заболевание проявляется прогрессирующей неврологической деградацией со спастичностью и мозжечковыми и экстрапирамидными знаками, такими как дистония (Nardocci et al., 1993). Может выявляться атрофия зрительного нерва и симптом вишневой косточки, акустическая реакция испуга встречается в половине случаев, несмотря на позднее появление данного симптома. Поражается тот же ген, что и при GM1 ганглиозидозе 1 типа, но отмечается гетерозиготность двух отдельных мутаций, одна их которых часто обнаруживается у гомозигот по GM1 ганглиозидозу 1 типа.
3. GM1 ганглиозидозы 3 типа (так называемые «взрослые») встречаются редко. Заболевание может начинаться в детстве или в подростковом возрасте в виде аномальной походки и ухудшения речи (Tanaka et al., 1995; Muthane et al., 2004). Проявления имеют вариабельный характер, часто встречаются дистония и паркинсонизм (Roze et al., 2005). Возможны проявления спиноцеребеллярной дегенерации. Варианты клинической картины включают дистонические формы (Tanaka et al., 1995) и атипичные случаи, например, с миопатией и кардиомиопатией (Charrow и Hvizd, 1986). Расстройство интеллекта при таких формах заболевания может наступать поздно или отсутствовать. Зарегистрировано снижение сигнала в области бледного шара и скорлупы на МРТ в T1-режиме (Tanaka et al., 1995), также отмечается лейкоэнцефалопатия (van der Voorn et al, 2004).
Диагностика GM1 ганглиозидозов облегчается при отсутствии экскреции с мочой мукополисахаридов, наличии костных аномалий, особенно поясничных позвонков, и пенистых макрофагов в костном мозге. Диагноз подтверждается отсутствием или заметным снижением активности бета-галактозидазы в лейкоцитах или фибробластах. При поздних формах заболевания (3 тип) отмечается частичный дефицит бета-галактозидазы (5-20% от нормы). Пренатальная диагностика может проводиться в культуре амниотических клеток или трофобласте.
Дефицит бета-галактозидазы отмечается также при болезни Моркио В типа, которая является заболеванием скелета без поражения нервной системы, и при галактосиалидозе, который описан вместе с сиалидозами.
в) GM3 ганглиозидозы являются исключительным состоянием, характеризуемым плохим физическим развитием, аномалиями лица, быстрой неврологической деградацией и ранней смертью. Существование и нозологическое положение данного заболевания остается неуточненным.
г) Дефицит лизосомальной альфа-N-ацетилгалактозиминидазы (рассмотрено в отдельной статье на сайте) может по патологическим проявлениям напоминать нейроаксональную дистрофию.