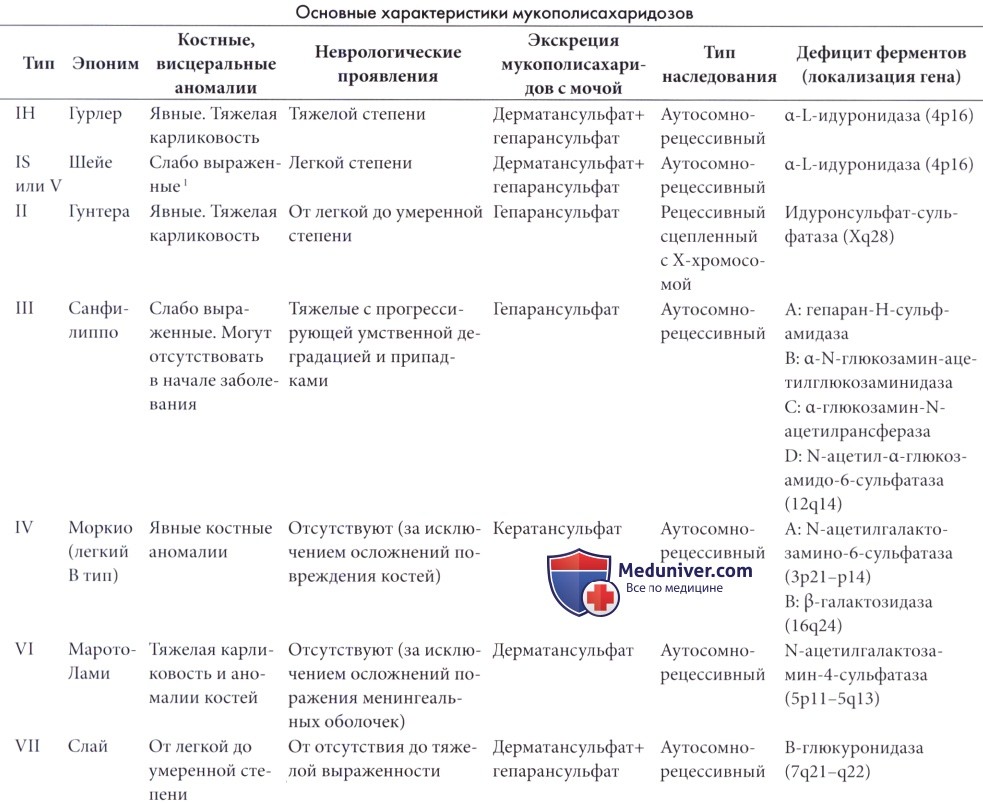
Мукополисахаридозы являются врожденными нарушениями метаболизма, вызванными дефицитом лизосомальной глюкозидазы или сульфатазы, который приводит к накоплению мукополисахаридов или гликозоаминогликанов в лизосомах. В таблице ниже приведены основные характеристики заболеваний данной группы.
Данные нарушения будут рассмотрены кратко, так как неврологические проявления часто отодвигаются на задний план дисморфическими и висцеральными проявлениями. Подробности можно узнать в специализированной литературе.
а) Болезни Гурлер и Шейе (мукополисахаридозы IH и 1-H/S типов). При болезни Гурлер широко распространены поражения внутренних органов, а клетки, содержащие полисахариды, обнаруживаются в ретикулоэндотелиальной системе и соединительной ткани.
Отмечается выраженное поражение центральной нервной системы в виде инфильтраций менингеальных оболочек, приводящих к гидроцефалии. Нейроны растянуты включениями, которые при электронной микроскопии имеют вид «телец зебры».
В головном мозге накопления представлены в основном GM2 и GM3 ганглиозидами, в то время как в периферических тканях основные скопления состоят из сульфата дерматана и гепарана, в большом количестве экскретирующихся с мочой.
Болезнь Гурлер является аутосомно-рецессивным заболеванием, которое встречается с частотой 1 на 100000 новорожденных. Больные дети кажутся нормальными при рождении, но развиваются медленно, костные изменения становятся явными в возрасте от шести месяцев до двух лет.
Выявляется гепатоспленомегалия, отделяемое из носа и пупочная грыжа. Характерные скелетные аномалии формируются постепенно вместе с огрублением черт лица, кифосколиозом, контрактурами суставов и расстройством интеллекта. Отмечаются помутнения роговицы, постоянным признаком является выраженная карликовость.
Голова большая, часто долихоцефалической формы. На КТ и МРТ выявляется расширение желудочков и пониженная плотность белого вещества полушарий (Zarifi et al., 2001; Matheus et al., 2004).
В области гипоталамуса часто выявляется кистозный арахноидит, вызывающий гидроцефалию. Расширение пространств Вирхова-Робена часто хорошо заметно на МРТ. Смерть наступает в возрасте до 20 лет.
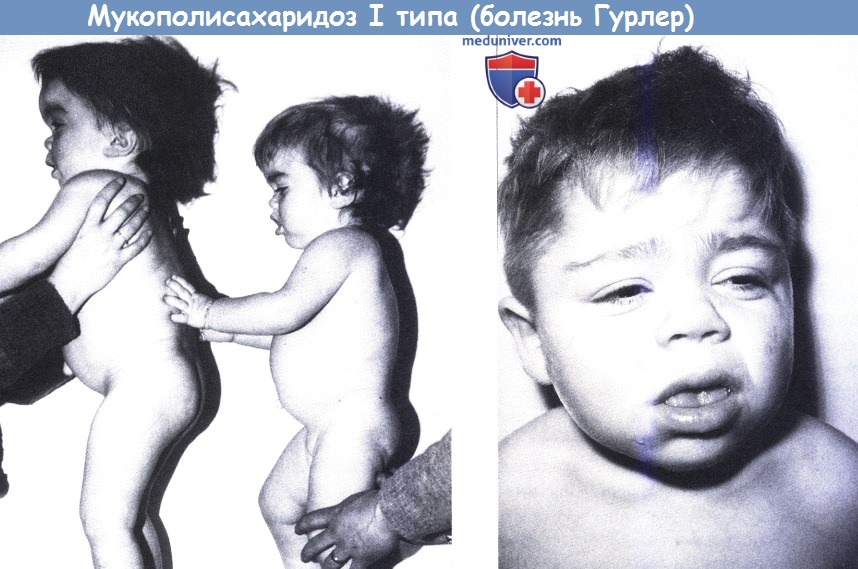
Мукополисахаридоз I типа (болезнь Гурлер).
Слева: Две сестры в возрасте 7 и 3 лет, отмечается увеличение головы со скафокефалией, типичный профиль и короткие кисти с широкими, частично согнутыми пальцами.
Справа: 12-летний мальчик с типичным лицом.
Диагноз может быть подтвержден L-идуронидазным исследованием. Наличие азурофильных гранул в гранулоцитах, гранул Рейли, вакуолизированных лимфоцитов (клетки Гассера I типа) и базофильных клеток в костном мозге (клетки Гассера II типа) является признаком, подтверждающим наличие заболевания. Возможна пренатальная диагностика.
Ген расположен на участке 4р16.3. Известно множество мутаций, две из которых являются причиной 80% случаев заболевания.
Лечение болезни Гурлер в настоящее время основано на ферментозаместительной терапии L-идуронидазой; данный метод является существенной заменой трансплантации костного мозга, которая была первым методом лечения с частичным успехом.
Ферментозаместительная терапия оказалась хорошо переносимой и эффективной в отношении висцеральных проявлений заболевания. Помутнения роговицы исчезают, а воздействие заболевания на рост и развитие принимает частичный характер. Раннее лечение необходимо до момента формирования стойких аномалий.
Уменьшения выраженности неврологических проявлений не отмечалось, тем не менее, описаны казуистические случаи повышения активности и восприимчивости у детей при раннем лечении (Wraith, 2005). Ограниченная эффективность объясняется неспособностью ферментов проникать через гематоэнцефалический барьер.
Попытки обойти барьер с помощью трансплантации гемопоэтических клеток или стволовых гемопоэтических клеток находятся на стадии изучения (Grewal et al., 2005; Lucke et al., 2007). В целом отмечается тенденция замещения трансплантации костного мозга ферментозаместительной терапией не только при мукополисахаридозах, но и при ряде других лизосомальных болезней накопления (Germain, 2005; Hoffmann и Mayatepek, 2005).
Данный метод лечения используется в основном при тяжелых состояниях с поражением центральной нервной системы, особенно при мукополисахаридозах I, II, III и VI типов. Предстоит решение ряда проблем, в частности, прохождения крупных молекул ферментов через гематоэнцефалический барьер. Изучаются и другие методы лечения, например, генная терапия.
Болезнь Гурлер-Шейе, официально известная как мукополисахаридоз V типа, характеризуется клинической картиной аналогичной болезни Гурлер и возможным заметным дисморфизмом (Schmidt et al., 1987), но поражение нервной системы ограничивается высокой частотой синдрома запястного канала, а умственные способности не изменяются. У некоторых пациентов развивается гидроцефалия.
б) Болезнь Гунтера (мукополисахаридоз II типа). Данное заболевание связано с дефицитом идуронат-2-сульфатазы (Kato et al., 2005) и наследуется сцепленно с Х-хромосомой. Заболевание встречается приблизительно в 5 раз реже, чем мукополисахаридозы I типа. В зависимости от наличия задержки умственного развития выделяются легкие и тяжелые формы. Неврологические аномалии при легких формах заболевания могут включать нейросенсорную тугоухость, пигментный ретинит и умеренную гидроцефалию (Froissart et al., 2002).
В результате утолщения соединительной ткани часто встречаются синдромы ущемления нервов. Определение уровня ферментов обеспечивает антенатальную и постнатальную диагностику. Ген расположен на участке Xq28, в 30% случаев отмечается его делеция. В тяжелых случаях возможно лечение с помощью трансплантации костного мозга.
в) Болезнь Санфипиппо (мукополисахаридоз III типа). Болезнь Санфилиппо является генетически гетерогенным заболеванием, известно четыре типа в зависимости от химического дефекта (табл. 8.1). Клинически заболевание проявляется прогрессирующей задержкой неврологического и умственного развития с началом в возрасте 2-6 лет. Помутнения роговицы отсутствуют, тем не менее, рано появляются аномально грубые черты лица и утолщение волос с последующим формированием неврологических нарушений. Часто встречается эпилепсия.
Течение заболевания неуклонно прогрессирующее, большинство пациентов умирает до 20 лет.
На начальных этапах диагностика затруднена в связи с преобладанием неврологических и психиатрических проявлений. Пациентам с необъяснимым регрессом показан плановый скрининг мочи на экскрецию му-кополисахаридов. Ферментное исследование позволяет проводить дифференцировку различных типов заболевания, так как фенотипические различия отсутствуют. Возможна пренатальная диагностика. Предпринимались попытки лечения с помощью трансплантации костного мозга, стволовых клеток и генной терапии, но результаты были в лучшем случае сомнительными.
г) Болезнь Моркио (мукополисахаридоз IV типа). Болезнь Моркио представлена двумя различными типами в зависимости от генетического и ферментативного дефекта. Клинические проявления представлены в основном костными аномалиями и помутнением роговицы без задержки умственного развития. При А типе заболевания постоянным признаком является гипоплазия зуба второго шейного позвонка, может отмечаться бульбоспинальная компрессия (Montano et al., 2003). Возможно повреждение менингеальных оболочек и нейронов. При легком течении заболевания или В типе неврологические проявления отсутствуют. Часто встречается атлантоаксиальная нестабильность, которая может быть причиной компрессии спинного мозга. Необходимо наблюдение и лечение у ортопеда и невропатолога.
д) Болезнь Марото-Лами (мукополисахаридоз IV типа) Данное заболевание характеризуется вариабельной тяжестью с различной степенью карликовости и поражения внутренних органов. Интеллектуальные нарушения имеют легкую степень или отсутствуют. Основным неврологическим осложнением является сдавление спинного мозга, обычно на уровне шеи, что может привести к квадриплегии. Это происходит в результате утолщения менингеальных оболочек в связи с накоплением мукополисахаридов и плохо поддается хирургическому лечению. Зарегистрированы исследования ферментозаместительной терапии (Karageorgos et al., 2004).
е) Болезнь Слая (мукополисахаридоз VII типа). Болезнь Слая является очень редким заболевание, которое может сопровождаться различными фенотипическими проявлениями от неиммунной водянки плода до случаев, имитирующих болезнь Гурлер с тяжелой задержкой неврологического развития, и случаев с ограниченными проявлениями без неврологических изменений (Bernsen et al., 1987; Saxonhouse et al., 2003).
ж) Другие формы мукополисахаридозов. Атипичные и неклассифицированные формы встречаются редко. Описан новый тип заболевания (Maroteaux, 1973) с основным проявлением в виде ацетоза в сочетании с экскрецией кератансульфата.