Болезнь Фабри сцепленное с полом заболевание связано с дефицитом (отсутствием, низкой активностью или быстрым катаболизмом) А изофермента альфа-галактозидазы (церамидтригексозидазы), вызванным несколькими мутациями GLA-гена, расположенного на Xq22 хромосоме. В результате в различных органах, особенно в почках, накапливается большое количество тригексозида.
Пенистые клетки с вакуолизированной цитоплазмой обнаруживаются в гладкой и поперечнополосатой мускулатуре, в костном мозге и почечных клубочках. В центральной нервной системе накопление происходит в стенках кровеносных сосудов и (в меньшей степени) автономной нервной системе. При МРТ можно выявить усиление перивентрикулярного сигнала и дискретные повреждения, позволяющие предположить демиелинизацию.
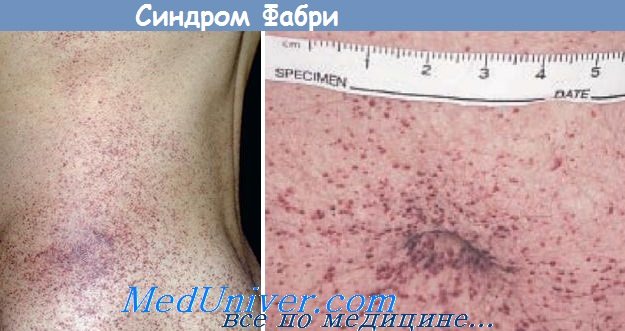
Клинические проявления заболевания обычно появляются в детстве. Аномалии кожи в форме точечных ангиоэктатических повреждений (angiokeratoma corporis diffusum) могут быть одним из характерных проявлений, которое часто обнаруживается на половых органах, в пупке или над бедрами, но редко встречаются на лице. Часто первым симптомом являются эпизоды боли и дизестезии в конечностях и иногда в области живота.
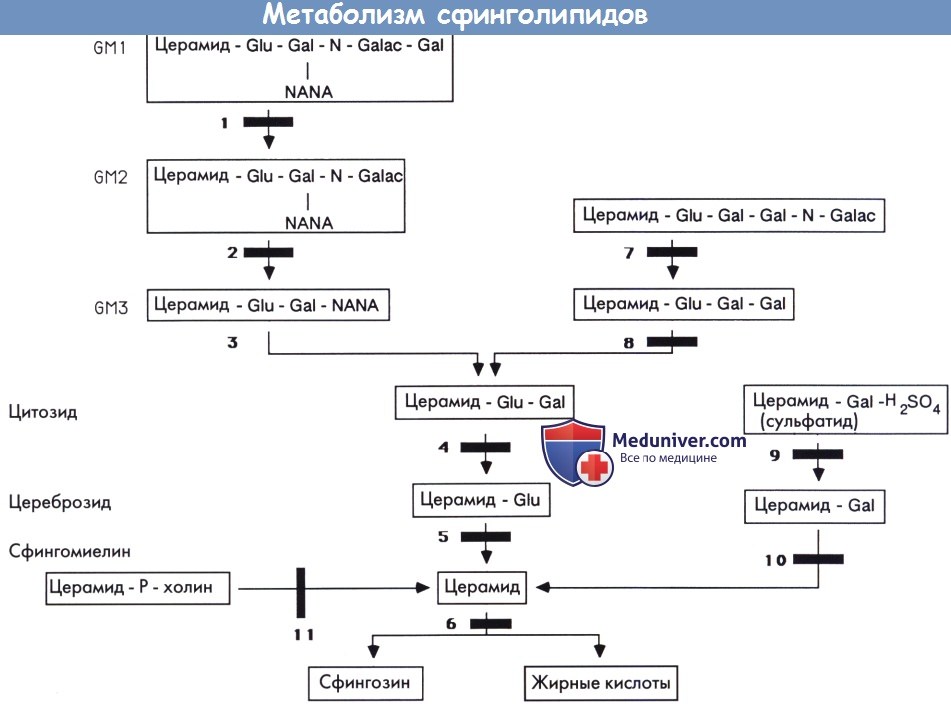
Метаболизм сфинголипидов.
Gal—галактоза. Glu—глюкоза. N-Galac—N-ацетилгалактозамин. NANA—нейраминовая кислота (N-ацетилнейраминовая кислота).
1 — β-галактозидаза. 2 — β-гексозаминидаза А (болезнь Тея-Сакса) и другие ганглиозидозы типа II GМ2.
3 — GM3 сиалидаза. 4 — лактозилцекамидаза (лактозилцерамидоз). 5 — β-глюкозидаза (глюкоцереброзидаза) (болезнь Гоше).
6 — церамидаза (болезнь Фарбера). 7 — β-гексозаминидаза В (болезнь Сандгоффа и другие О варианты GM2 ганглиозидозов).
8 — церамид-тригексозидаза/α-галактозидаза (болезнь Фабри). 9 — цереброзидсульфатаза/арилсульфатаза* (метахроматическая лейкодистрофия).
10 — β-галактозидаза/β-галактоцереброзидаза (болезнь Краббе). 11 — сфингомиелиназа (болезнь Ниманна-Пика А и В).
*Активность, оцененная на искусственных субстратах, не всегда соответствовала активности цереброзиодсульфатазы.
Боль имеет глубокий, жгучий характер и возникает в виде эпизодов, длящихся от нескольких часов до недель. Данный симптом болезни Фабри часто сочетается с необъяснимой лихорадкой. При осмотре в щелевой лампе видна cornea verticillata, которая часто отмечается у гетерозигот (Watts и Gibbs, 1986). Зарегистрированы случаи периферической нейропатии, часто отмечается ангидроз. Приблизительно у 30% пациентов выявляются пороки сердца, включая пролапс митрального клапана и кардиомиопатию, часто развиваются транзиторные ишемические атаки (Ries et al., 2005).
Заболевание характеризуется прогрессирующим течением, изменения со стороны центральной нервной системы и внутренних органов часто проявляются очаговыми неврологическими симптомами, гипертензией и инфарктом миокарда. Обычно причиной смерти является поражение почек. У женщин-носительниц обычно отмечается позднее начало и меньшая выраженность симптомов.
Ранее подтверждение диагноза возможно путем выявления альфа-галактозы в плазме, лейкоцитах или фибробластах. Возможна антенатальная диагностика и выявление носителей.
Лечение болезни Фабри включает предотвращение болевых эпизодов, обычно с помощью карбамазепина, габапентина или фенитоина, и терапию почечной недостаточности. Трансплантация почек оказывает незначительное воздействие на поражение центральной нервной системы, хотя возможно уменьшение выраженности сенсорных симптомов и улучшение функции почек. Ферментозаместительная терапия изменила развитие заболевания.
В ходе крупных контролируемых исследований больных мужчин была продемонстрирована эффективность и безопасность рекомбинантных ферментных препаратов, которые уменьшают боль и стабилизируют функцию почек (Clarke и Iwanochko, 2005; Schaefer et al., 2005). Качество жизни пациентов значительно улучшилось (Hoffmann et al, 2005). Профилактическое лечение женщин-носительниц без появления симптомов не рекомендуется. Выявлено образование антител к ферментам (Linthorst et al., 2004), но их клиническая значимость не ясна.