Центральная диффузная миелинопатия (MCD, болезнь исчезающего белого вещества (VWMD), детская атаксия с гипомиелинизацией ЦНС, (САСН)) - причины, диагностика, лечение
В результате систематического анализа прогрессирующих заболеваний белого вещества с использованием современных радиологических и генетических методик среди наследственных заболеваний белого вещества выявлен ряд «новых» нозологических единиц, многие из которых были описаны ранее в единичных отчетах.
Центральная диффузная миелинопатия (MCD); болезнь исчезающего белого вещества; детская атаксия с гипомиелинизацией ЦНС (САСН) заболевание, называемое в настоящее время болезнь исчезающего белого вещества (VWMD), представляет собой наиболее распространенное наследственное заболевание белого вещества у детей (van der Knaap et al., 2006). После упоминаний в ранних отчетах заболевание было описано в 1993 г. Hanefeld et al. как диффузное поражение белого вещества с общими проявлениями на МРТ и протонной МР-спектроскопии. Авторы описали трех детей, включая брата с сестрой, у которых развилась атаксия в результате минимальной травмы головы или неспецифической инфекции. У третьего пациента (мальчика) атаксия развилась после минимальной травмы головы и сохранялась в течение трех дней. На КТ, выполненном в эти дни, видны диффузные изменения белого вещества.
У мальчика отмечалось начальное восстановление, но атаксия возобновлялась после минимальной травмы головы или неспецифического заболевания, сопровождавшегося лихорадкой. В возрасте четырех лет у него отмечалась спастично-атаксическая походка, активные рефлексы, клонус и двусторонний рефлекс Бабинского. В течение следующих месяцев и лет у пациента развилась атрофия зрительного нерва, нарушения глотания и большие судорожные припадки. Пациент умер от бронхопневмонии. На МРТ обоих сибсов отмечены практически идентичные диффузные изменения белого вещества. МР-спектроскопия, выполненная одновременно, выявила полную утрату метаболитов в пораженном белом веществе, в то время как в сером веществе отмечалось только легкое, но несомненное снижение уровня NNA.
Вид белого вещества на МР-спектроскопии напоминал спинномозговую жидкость с заметным повышением уровня лактата. В последующие годы было описано большое количество сходных случаев (Schiffmann et al., 1994; van der Knaap et al., 1997). Leegwater et al. (2001) and van der Knaap et al. (2002a) продемонстрировали мутации во всех субъединицах эукариотического транслокационного фактора eIF2B. Большая часть мутаций наследуется рецессивно. Доминантное наследование предполагается в отношении семейства мутаций, зарегистрированных Labauge et al., (2005). В ходе дальнейших исследований выявлено, что варианты заболевания с ранним началом, включая лейкоэнцефалопатию Кри (Black et al., 1988), вызваны мутацией eIF2B5 (Fogli et al., 2002a, b). Данная мутация также описана при других видах заболевания с тяжелым летальным началом лейкоэнцефалопатии в антеи перинатальном периоде. Кроме того, другие аномалии часто представлены маловодием, внутриутробной задержкой роста, катарактой, панкреатитом, гепатоспленомегалией, гипоплазией почек и дисгенезом яичников (van der Knaap et al., 2003a). Сочетание поражения белого вещества с дисгенезом яичников описаны Schiffmann et al. (1997). В настоящее время ясно, что VWMD характеризуется широким спектром клинических проявлений, включая рано и остро начинающиеся формы (классический VWMD/MCD синдром) и более легкие формы с началом во взрослом возрасте (Fogli и Boespflug-Tanguy, 2006; van der Knaap et al., 2006).
Несмотря на большое количество пациентов с описанными мутациями, взаимосвязь генотипа и фенотипа при поражении гена eIF2B до сих пор не ясна.
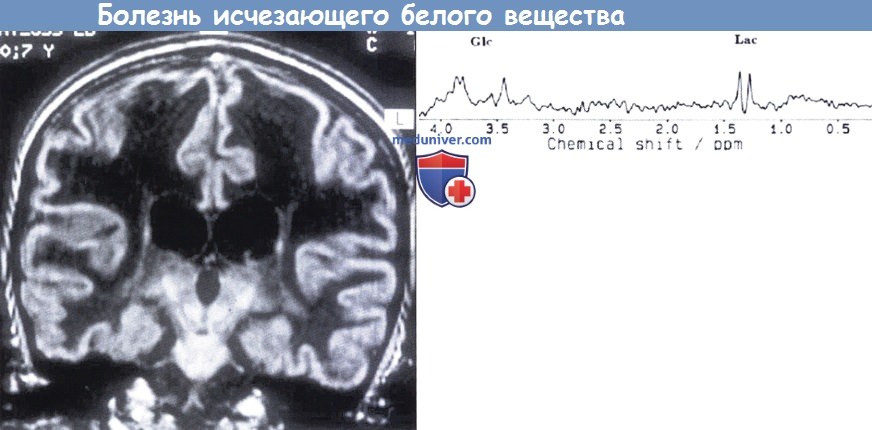
Болезнь исчезающего белого вещества.
На МРТ (вверчу) видно практически полное исчезновение белого вещества обоих полушарий с сигналом, подобным сигналу спинномозговой жидкости.
На МР-спектроскопии (внизу) видно практически полное отсутствие нормальных пиков.
При нейропатологическом обследовании были выявлены зависимые от возраста аномалии. В тяжелых случаях описываются кистозные полости. При микроскопическом исследовании отмечается утрата миелина, но воспалительная реакция отсутствует. Серое вещество в целом сохранено, но отмечается полная утрата аксонов в области кист. При продолжительном течении заболевания выявляется серьезное поражение олигодендроцитов (Rodriguez et al. 1999; van Haren et al., 2004). По результатам недавнего исследования культуры клеток головного мозга мальчика, умершего в возрасте 12 лет, в условиях in vitro выявлено быстрое образование олигодендроцитов, но отмечалось выраженное аномальное снижение выработки GFAP-позитивных астроцитов. Дефицит нормально функционирующих астроцитов может воздействовать на патогенез VWMD.
Диагностика классического типа VWMD/MCD основана на клинической картине и течении заболевания: нормальное развитие в течение первых 2-3 лет с последующим появлением атаксии и спастичности (обычно после легкой травмы или неспецифической болезни, сопровождающейся лихорадкой), быстрым прогрессированием с утратой моторных функций, припадками и поздним формированием задержки умственного развития. Признаки периферической нейропатии отсутствуют. В альтернативных случаях возможно частичное восстановление с последующими рецидивами, приводящими к прогрессивному ухудшению.
На МРТ выявляется диффузное поражение белого вещества с характерным ранним вовлечением центрального тракта покрышки. На МР-спектроскопии подтверждается утрата миелина, имеющая метаболическое происхождение, отличающаяся от спинномозговой жидкости только наличием лактата. Заболевание прогрессирует до формирования тяжелой инвалидности, большинство детей с классическим фенотипом заболевания умирает до 20-летнего возраста.
Повышение уровня глицина и снижение концентрации азиалотрансферрина описано в качестве возможного маркера заболевания (van der Knaap et al., 1999). Тем не менее MPT-критерии, сформулированные van der Knaap, используются для диагностики чаще всего. Ввиду широкой фенотипической вариабельности поражения eIF2B диагноз должен подтверждаться генетическим анализом.
Возможна пренатальная диагностика поражения eIF2B. Лечение в настоящее время отсутствует.