2. Определения:
• Группа редких остеохондродисплазий, для которых характерны укорочение трубчатых костей, короткие горизонтальные ребра, деформация грудной клетки, ± полидактилия, ± аномалии внутренних органов:
о Подтипы (АТД 1-14; СКРП 1—4) отличаются характером поражения внутренних органов и строением метафизов, генетически гетерогенны, имеют общие фенотипические проявления
о Принадлежат к группе аутосомно-рецессивных цилиопатий со скелетными нарушениями
б) Лучевая диагностика:
1. Общие сведения:
• Критерии диагностики:
о Триада: микромелия, полидактилия, короткие горизонтальные ребра
о Диагностируется с 15-16 нед. на основании микромелии и выраженного укорочения ребер
о В I триместре при беременности высокого риска - утолщение воротникового пространства
• Асфиктическая торакальная дисплазия ± полидактилия (АТД типа 1,3): синдром Жена:
о Вытянутая узкая грудная клетка с короткими горизонтальными ребрами
о Кистозная дисплазия почек
о Полидактилия встречается реже (14%)
о Трубчатые кости поражены в меньшей степени, большеберцовые кости не изменены
о Часто - смерть в перинатальном периоде вследствие тяжелой дыхательной недостаточности
о У пациентов, переживших детство, обычно развиваются болезни почек и печени
о АТД типа 1 связана с мутациями на 15q13
о АТД типа 3 связана с мутациями гена DYNC2H1
• Асфиктическая торакальная дисплазия ± полидактилия (АТД типа 3): синдромы Салдино-Нунана (СКРП типа 1) и Верма-Наумова (СКРП типа 3) имеют одинаковые проявления:
о Постаксиальная полидактилия
о Водянка плода
о Дефекты перегородок сердца, коарктация аорты, транспозиция магистральных сосудов
о ЖКТ/мочеполовая система: кисты почек, клоакальные аномалии, атрезия влагалища, влагалищные свищи, атре-зия ануса
о Перинатальная летальность
о Рентгенологические признаки: гипоплазия подвздошных костей с уплощением крыш вертлужных впадин, округлые позвонки с передними расщелинами, трубчатые кости с заостренными эпифизами/вогнутыми диафизами, латеральные остеофиты метафизов, зазубренные эпифизы, при СКРП типа 1 - отсутствие малоберцовой кости
о При СКРП типа 3 аномалии внутренних органов встречаются реже
о АТД типа 3 вызвана мутациями гена DYNC2H1
• Асфиктическая торакальная дисплазия ± полидактилия (АТД типа 6): синдром Маевского (СКРП типа 2):
о Пре- и постаксиальная полидактилия
о Водянка плода
о Орофациальные расщелины, часто по срединной линии
о Гениталии промежуточного типа
о Аномалии ЦНС
о Рентгенологические признаки: короткие горизонтальные ребра, укороченные трубчатые кости со сглаженными эпифизами, укороченные большеберцовые кости (короче малоберцовых) овоидной формы, подвздошные кости не изменены
о АТД типа 6 связана с мутациями гена NEK1
• Асфиктическая торакальная дисплазия грудной клетки ± полидактилия (АТД типа 12): синдром Бимер-Лангера (СКРП типа 4):
о В 50% случаев - пре- и постаксиальная полидактилия
о Аномалии внутренних органов: омфалоцеле, пороки сердца, кистозная дисплазия или гипоплазия почек, дольчатый язык с короткой уздечкой, гениталии промежуточного типа
о Срединная расщелина лица о ЦНС: гидроцефалия, ГПЭ, гамартомы
о Кистозная гигрома в I триместре
о Рентгенологические признаки: короткие горизонтальные ребра, уменьшенные подвздошные кости, укороченные трубчатые кости со сглаженными метафизами, искривление локтевых и лучевых костей
о В небольшой выборке пациентов мутации генов NEK1 и DYNC2H1 отсутствовали
• В других небольших сериях случаев диагностированы АТД типа 2, 4, 5, 7—11, 13, 14 с множеством общих признаков
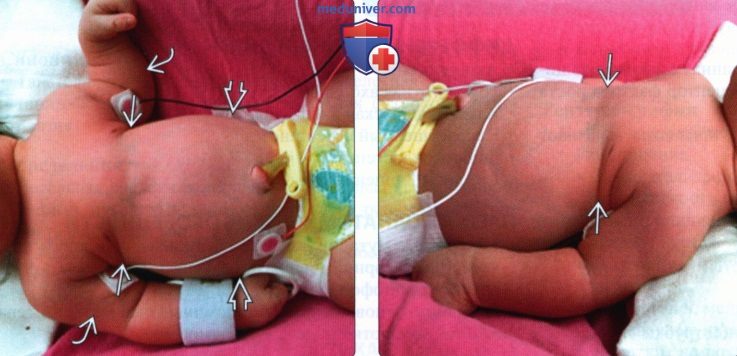
(Слева) Новорожденный с синдромом Жена (АТД типа 1). Грудная клетка предельно уменьшена, сужена, контрастирует с выпуклым животом, кожа будто натянута в складку. Конечности укорочены.
(Справа) Тот же случай. Новорожденный с синдромом Жена, вид сбоку. Грудная клетка значительно сужена. Малые размеры грудной клетки приводят к гипоплазии легких, перинатальная смертность при данном заболевании достигает 70%. Ребенок умер вскоре после рождения.
2. УЗИ при синдроме коротких ребер-полидактилии у плода:
• Уменьшение грудной клетки, укорочение ребер:
о Окружность грудной клетки <5-го процентиля при нормальной ОЖ
• Возможна кистозная дисплазия почек
• Внескелетные аномалии: пороки сердца и гениталий, орофациальные расщелины
• Оссификация костей в норме
• Укорочение трубчатых костей:
о Возможно незначительное искривление, переломы отсутствуют
• Утолщение воротникового пространства в I триместре
• При тяжелом поражении почек возможно маловодие
• Постаксиальная полидактилия (не всегда)
3. Рентгенография при синдроме коротких ребер-полидактилии у плода:
• Рентгенография новорожденного:
о Ребра укорочены, горизонтальные, грудная клетка уменьшена
о Укорочение подвздошных, седалищных и лобковых костей
о Медиальные и латеральные остеофиты подвздошных костей
о Укорочение, искривление конечностей
о Конусовидные эпифизы
• Нарушения наиболее выражены в младенчестве, у выживших грудная клетка изменена в меньшей степени
4. Рекомендации по лучевой диагностике:
• При повышенном риске заболевания - ТВУЗИ
• 3D/4D УЗИ во II и III триместрах
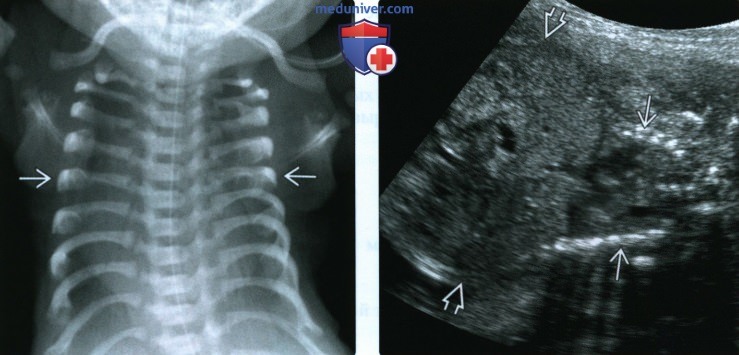
(Слева) Рентгенограмма новорожденного с синдромом Жена, прямая проекция. Узкая грудная клетка. Ребра короткие, прямые оссификация в норме. Сердце выглядит увеличенным из-за малых размеров грудной, клетки.
(Справа) УЗИ плода во II триместре. Синдром Жена, АТД типа 1 (подтверждена в постнатальном периоде). По сравнению с животом грудная клетка значительно сужена. ОЖ соответствует 55-му процентилю, окружность грудной клетки менее 5-го процентиля для данного ГВ.
в) Дифференциальная диагностика синдрома коротких ребер-полидактилии у плода:
1. Синдром Эллиса-ван Кревельда:
• Хондроэктодермальная дисплазия
• Редкое заболевание, чаще встречается среди амишей
• Поражение грудной клетки менее выраженное, укорочение трубчатых костей
• Неполная срединная расщелина верхней губы
• В 60% случаев - пороки сердца (дефекты перегородок, единое предсердие)
• Постаксиальная полидактилия
• Интеллект не страдает
• Мутации генов EVC1 и EVC2 на 4р 16; аутосомно-рецессивный тип наследования
2. Синдром Мора-Маевского:
• Оро-фацио-дигитальный синдром типа 4
• Дифференциальная диагностика между СКРП и оро-фациодигитальным синдромом типа 5 затруднена:
о Могут представлять единый спектр
• Тяжелое поражение большеберцовых костей, более длинные ребра
• Неабсолютная летальность новорожденных
3. Синдром Барнса:
• Гипоплазия грудной клетки и тазовой кости, стеноз гортани
• Укорочение ребер менее выражено, чем при синдроме Жена
• Остеофиты подвздошных костей отсутствуют, почки не поражены
• Наследуется по аутосомно-доминантному типу
4. Однородительская дисомия 14 (отцовская)/синдром Кагами-Огата:
• Характерный фенотип с уменьшенной колоколообразной грудной клеткой
• При рентгенографии - характерный признак «вешалок» (передние части ребер изогнуты каудально)
• Нарушения локализуются в метилированном участке хромосомы 14q
• Аномалии челюстно-лицевой области
• Дефекты брюшной стенки
• Плацентомегалия
• Многоводие
5. Синдром Сенсенбреннера (краниоэктодермальная дисплазия):
• Редкая цилиопатия с поражением скелета и аутосомно-рецессивным типом наследования
• Сагиттальный краниосиностоз
• Характерные черты лица
• Карликовость
• Узкая грудная клетка
• Признаки эктодермальной патологии: редкие волосы, гиподонтия, микродонтия
• Установлена связь с мутациями четырех генов: WDR35, IFT122, IFT43, WDR19
6. Синдром Майнцера-Сальдино:
• Цилиопатия с поражением скелета
• Конусовидные эпифизы
• Ранняя ретинодистрофия тяжелой степени
• Хроническая почечная недостаточность, достигающая терминальной стадии в подростковом возрасте
• Мутации гена IFT172, кодирующего белок интрафлагеллярного транспорта
• Как правило, возникает случайно
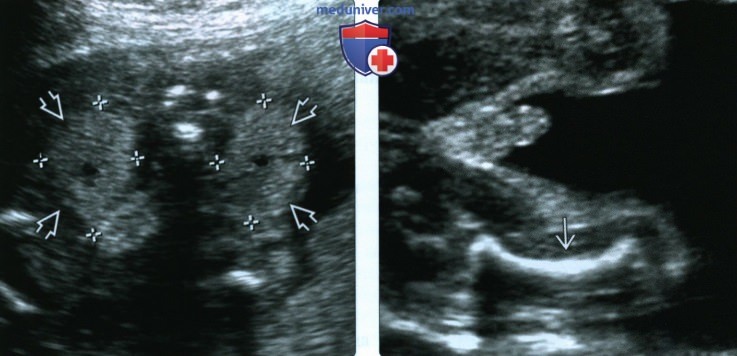
(Слева) УЗИ плода во II триместре с подозрением на синдром Жена, поперечная плоскость. Обе почки увеличены, гиперэхогенные, что позволяет заподозрить кистозную дисплазию. Данная находка характерна для нескольких типов синдрома коротких ребер - полидактилии; в тяжелых случаях может развиться маловодие.
(Справа) УЗИ плода мужского пола во II триместре. Синдром Жена. Бедренная кость укорочена, искривлена, оссификация в норме. При данном заболевании нередко наблюдают укорочение и умеренное искривление трубчатых костей.
г) Патологоанатомические особенности:
1. Общие сведения:
• Этиология:
о Первичная цилиарная дискинезия с поражением хондроцитов, в том числе дыхательных путей
о Мутации гена DYNC2H1, кодирующего компонент динеинового комплекса цитоплазмы, который участвует в формировании ресничек (цилиопатия); установлена связь с мутациями некоторых других генов
о Скелетные проявления связаны с патологией сигнального пути Sonic hedgehog в ресничках хондроцитов:
- Нарушение организации микротрубочек в цитоскелете хондроцитов
• Генетические факторы:
о Аутосомно-рецессивный тип наследования (почти всегда)
• Сопутствующие аномалии:
о Различные поражения внутренних органов:
- Фиброз печени и поджелудочной железы, дистрофия сетчатки, пороки сердца, орофациальные расщелины
о Высокий риск смерти в перинатальном периоде вследствие сужения грудной клетки, а также печеночной, почечной или сердечной недостаточности
2. Микроскопические изменения:
• Нарушение баланса между резорбцией хряща и остеогенной дифференцировкой во всех зонах роста
• Неправильная, «рваная» энхондральная оссификация
• Гипоплазия легких со значительным снижением числа альвеол
• При синдроме Жена - кистозная дисплазия почек и перипортальный фиброз печени
д) Клинические особенности:
1. Клиническая картина:
• Самые частые субъективные и объективные симптомы:
о Высокая вариабельность фенотипа
о Вытянутая и суженная грудная клетка
о Брахидактилия, укорочение конечностей
о Кистозная дисплазия почек
о Постаксиальная полидактилия (не всегда)
• Другие субъективные и объективные симптомы:
о Тяжелая дыхательная недостаточность у новорожденных вызвана сужением грудной клетки и подскладочным стенозом гортани вследствие нарушения развития дыхательных путей
2. Демографические особенности:
• Эпидемиология:
о Встречается редко - 1:70 000 родов
3. Естественное течение и прогноз:
• В 70% случаев — смерть в нео- или постнатальном периоде вследствие гипоплазии легких
• Выживают в связи с увеличением грудной полости
• Нетяжелые случаи проявляются карликовостью ± почечной недостаточностью в детстве
• При синдроме Жена почечная недостаточность достигает терминальной стадии в старшем детском возрасте
• Тяжелое поражение печени → билиарный цирроз → портальная гипертензия
4. Лечение синдрома коротких ребер-полидактилии у плода:
• Показано генетическое консультирование
• Возможно прерывание беременности
• Для расчета риска повторного возникновения принципиальную роль играет подтверждение диагноза у новорожденного
• Операции по расширению ребер и грудной клетки
• Урсодезоксихолевая кислота для стабилизации клеточных мембран гепатоцитов
• Трансплантация почки
е) Список использованной литературы:
1. Mei L et al: Targeted next-generation sequencing identifies novel compound heterozygous mutations of DYNC2H1 in a fetus with short rib-polydactyly syndrome, type III. Clin Chim Acta. 447:47-51, 2015
2. Schmidts M: Clinical genetics and pathobiology of ciliary chondrodysplasias. J Pediatr Genet. 3(2):46-94, 2014
3. Baujat G et al: Asphyxiating thoracic dysplasia: clinical and molecular review of 39 families. J Med Genet. 50(2):91-8, 2013
4. Huber C et al: WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-кВ pathway in cilia. Am J Hum Genet. 93(5):926-31, 2013