б) Лучевая диагностика. УЗИ при синдроме Фрейзера:
• Глазные яблоки уменьшены или отсутствуют:
о Анофтальм, микрофтальм, криптофтальм
о Исследование выполняют в поперечной плоскости на уровне глаз:
- Оценивают костные стенки глазниц и глазные яблоки
о Измеряют диаметр глазницы (ДГ) и соотносят значение с нормативными данными
о Нормальные значения соответствуют «правилу третей»:
- Интраорбитальный размер = ДГ:
Между внутренними стенками глазниц должен помещаться «третий глаз»
Увеличение интраорбитального размера связано либо с уменьшением размеров глазных яблок (синдром Фрейзера), либо с гипертелоризмом
• Аномалии развития мочевыводящих путей:
о Агенезия или гипоплазия почек
о МКДП
• Синдактилия
• Аномалии развития гортани и трахеи:
о Могут вызывать сужение и последующую обструкцию верхних дыхательных путей:
- Легкие увеличены, гиперэхогенны
- Эвентрация диафрагмы
- Сдавление сердца
- Характерен асцит, иногда выраженный
• Маловодие вследствие пороков развития почек
• 3D УЗИ позволяет исследовать черты лица, однако возможности метода нередко ограничены маловодием
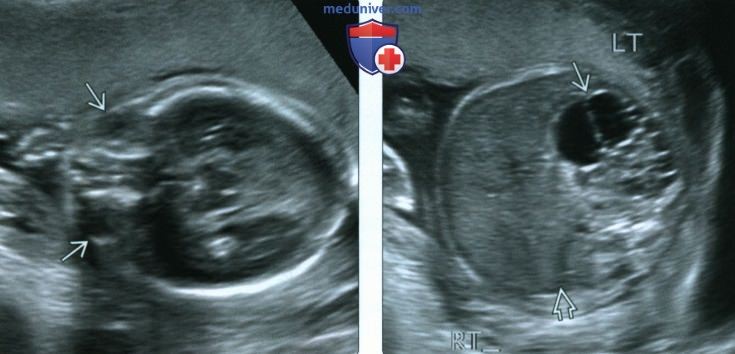
(Слева) УЗИ плода в 20 нед., поперечная плоскость. Срез через глазницы. Определяется микрофтальм. Межглазничное расстояние кажется достаточно большим (в норме это расстояние соответствует ширине одного глазного яблока), что объясняется уменьшением размеров глазных яблок, как в данном случае, или гипертелоризмом.
(Справа) Тот же плод. УЗИ брюшной полости. Визуализируется мультикистозная дисплазированная левая почка в правом почечном ложе почка отсутствует. Сочетание пороков развития почек и органа зрения позволяет заподозрить синдром Фрейзера.
в) Дифференциальная диагностика синдрома Фрейдера у плода:
1. Анофтальм, микрофтальм:
• Т13
• Синдром Уокера-Варбург: врожденная мышечная дистрофия в сочетании с пороками развития головного мозга и органа зрения:
о Первичное персистирующее гиперпластическое стекловидное тело имеет вид гиперэхогенных полос или объемного образования внутри глазницы
• Синдром CHARGE: колобома, порок сердца, атрезия хоан, задержка умственного развития, аномалии гениталий и органа слуха
• Аномалии развития органа зрения, вызванные мутацией гена SOX2
2. CAKUT:
• Аномалии развития мочевыводящих путей часто обозначают термином «врожденные аномалии почек и мочевыводящих путей» (congenital anomalies of kidney or urinary tract)
• CAKUT в сочетании с внепочечными проявлениями характерны для многих синдромов, отдельные признаки которых нередко совпадают
• Синдром Вольфа-Хиршхорна, или синдром делеции 4р: делеция дистального конца короткого плеча хромосомы 4; лицевая дизморфия, задержка роста и умственного развития, CAKUT
• Гипоплазия почек: ВМР4, SIX2\ микрофтальм, расщелина верхней губы, гипоплазия или дисплазия почек
• Синдром Барде-Бидля: многие гены, кодирующие функциональные протеины цилий; ретинопатия, аномалии развития пальцев, ожирение, мужской гипогонадизм, дисплазия почек
• Синдром Смита-Лемли-Опица: ген, кодирующий фермент, необходимый для биосинтеза холестерина (DHCR7); лицевая дизморфия, микроцефалия, синдактилия, нарушение умственного развития, дисплазия почек
• Бранхио-ото-ренальный синдром: EYA1, SIX1, SIX5, MYOG; тугоухость, боковые кисты шеи, CAKUT
• Колобома-ренальный синдром: РАХ2; колобома сосудистой оболочки глаза, CAKUT
(Слева) Тот же плод. УЗИ грудной клетки, фронтальный срез. Легкие расширены и гиперэхогенны вследствие врожденной обструкции верхних дыхательных путей. Диафрагма уплощена. Также определяется маловодие, связанное с наличием аномалий развития почек.
(Справа) МРТ плода в 27 нед., Т2-ВИ, поперечная плоскость. Выраженный двусторонний микрофтальм является одной из отличительных черт синдрома Фрейзера.
г) Патологоанатомические особенности. Общие сведения:
• Генетические факторы:
о Аутосомно-рецессивный тип наследования:
- Во многих случаях заболевание возникает в результате кровосмешения
о Мутации генов FRAS1, FREM2 и GRIP1:
- Все указанные гены участвуют во взаимодействии между зачатком мочеточника и метанефрогенной мезенхимой
д) Клинические особенности:
1. Клиническая картина:
• Самые частые субъективные и объективные симптомы:
о Наиболее явной патологией являются аномалии развития почек:
- Проводят тщательный поиск других патологий
• Для диагностики заболевания в постнатальном периоде оценивают наличие «больших» и «малых» критериев:
о «Большие» критерии:
- Криптофтальм
- Патология мочевыводящих путей
- Гениталии промежуточного типа
- Пороки развития гортани и трахеи
- Положительный семейный анамнез
о «Малые» критерии:
- Аноректальные аномалии
- Дисплазия ушных раковин
- Нарушения оссификации черепа
- Расщелина верхней губы и нёба
- Пупочная грыжа
3. Естественное течение и прогноз:
• Заболевание часто имеет летальный исход
• Продолжительность жизни зависит от количества и тяжести аномалий
• У многих выживших детей отмечаются задержка развития и низкий интеллект
е) Особенности диагностики. Признаки, учитываемые при интерпретации изображений:
• Пороки развития почек и мочевыводящих путей у плодов встречаются довольно часто и могут быть изолированными, однако следует всегда проводить поиск внепочечной патологии
• Патология почек и лица характерна для многих синдромов
ж) Список использованной литературы:
1. Ozemri Sag S et al: A novel mutation in the FRAS1 gene in a patient with Fraser syndrome. Genet Couns. 26(1):21—7, 2015
2. Kohl S et al: Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 25(9): 1917-22, 2014
3. Barisic I et al: Fraser syndrome: epidemiological study in a European population. Am J Med Genet A. 161A(5):1012-8, 2013
4. Mesens T et al: Congenital high airway obstruction syndrome (CHAOS) as part of Fraser syndrome: ultrasound and autopsy findings. Genet Couns. 24(4):367-71, 2013