Склерозирующие дисплазии костной ткани - кратко с точки зрения внутренних болезней
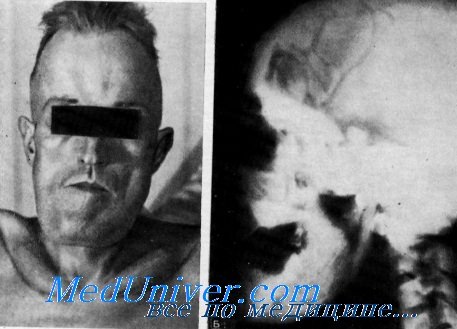
Склерозирующие дисплазии костной ткани - это редкие заболевания, характеризующиеся остеосклерозом и повышенным образованием кости. Болезнь ван Бухема и склеростеоз являются рецессивными нарушениями, вызванными мутациями с потерей функции в гене SOST, которые обычно подавляют формирование кости (см. рис. ниже).
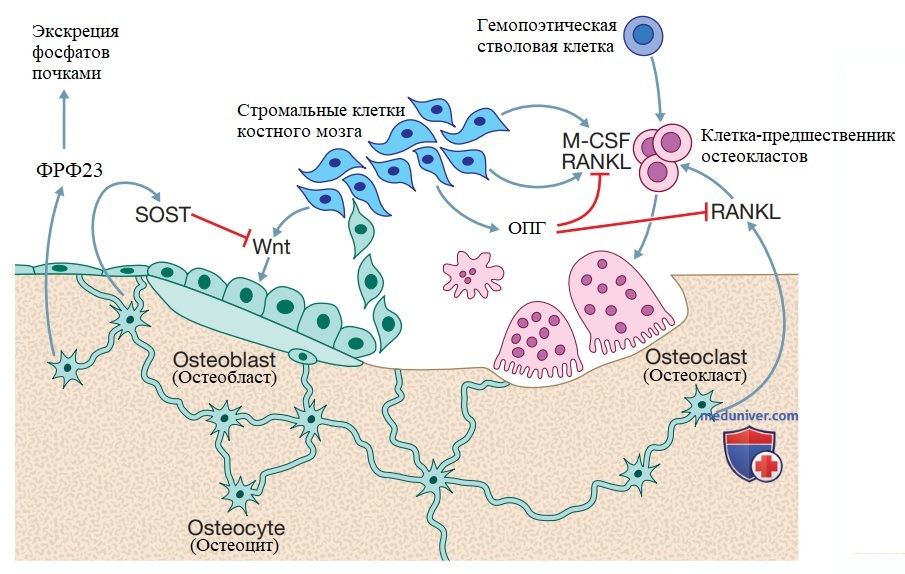
Клеточные и молекулярные регуляторы ремоделирования костной ткани. Клетки-предшественники остеокластов происходят из гемопоэтических стволовых клеток. Они дифференцируются в зрелые остеокласты в ответ на действие лиганда рецептора-активатора ядерного фактора каппа-В (RANKL), который продуцируется остеоцитами, стромальными клетками костного мозга, активированными Т-лимфоцитами (не изображены на рисунке) и макрофагальным колониестимулирующим фактором (М-КСФ), который вырабатывается стромальными клетками костного мозга. Остеопротегерин (ОПГ) — важный компонент костного гомеостаза, который подавляет резорбцию костной ткани остеокластами, блокируя эффект RANKL. Остеобласты, происходящие из стромальных клеток костного мозга, отвечают за формирование кости. Сигнальные молекулы семейства Wnt стимулируют активность остеобластов, а склеростин (SOST), продуцируемый остеоцитами, напротив, подавляет ее. В дополнение к своей роли в регуляции активности остеокластов и остеобластов, остео-циты выполняют эндокринную функцию в регуляции обмена фосфата, продуцируя фактор роста фибробластов 23 (ФРФ23), который стимулирует экскрецию фосфатов почками
В результате недостаток склеростина приводит к интенсификации образования костей и к их разрастанию, следствием чего является увеличение в размерах черепа и челюсти, высокий рост и паралич черепных нервов. Эффективное лечение отсутствует. Синдром высокой костной массы представляет собой доброкачественное заболевание, вызванное мутациями в генах LRP4 или LRP5 и характеризующееся необычно высокой плотностью кости.
Болезнь ван Бухема
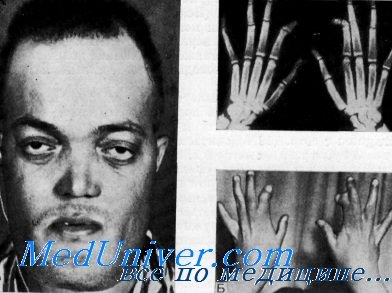
Склеростеоз
Мутации делают рецепторы LRP устойчивыми к ингибирующему действию SOST. Большинство пациентов не имеют симптомов, но в более позднем возрасте возможно образование нёбного нароста (torus palatinus) и увеличение в размерах нижней челюсти.
Лечение обычно не требуется. Болезнь Камурати—Энгельманна является аутосомно-доминантным заболеванием, вызванным усилением функции в гене TGFB1. Она сопровождается болями в костях, мышечной слабостью и остеосклерозом, которые преимущественно поражают диафиз длинных костей. При болях в костях могут помочь глюкокортикоиды, хотя обычно также необходимо применение анальгетиков.