Нарушения аминокислотного метаболизма - кратко с точки зрения внутренних болезней
Врожденные нарушения метаболизма аминокислот обычно проявляются в неонатальном периоде, и их лечение может быть пожизненным. Тем не менее некоторые нарушения, особенно связанные с транспортом аминокислот, могут манифестировать и в более позднем возрасте.
а) Фенилкетонурия. Фенилкетонурия наследуется по аутосомнорецессивному типу. Это заболевание вызвано мутациями с утратой функции в гене РАН, который кодирует фенилаланингидроксилазу, фермент, необходимый для расщепления фенилаланина. В результате фенилаланин в большом количестве накапливается в крови новорожденного, вызывая умственную отсталость.
Генетика фенилкетонурии
Диагноз «фенилкетонурия» почти всегда ставят с помощью рутинного скрининга новорожденных. Лечение включает пожизненное соблюдение диеты с низким содержанием фенилаланина. Ранние и адекватные диетические ограничения предотвращают развитие выраженной умственной отсталости, хотя может сохраняться небольшое снижение IQ.
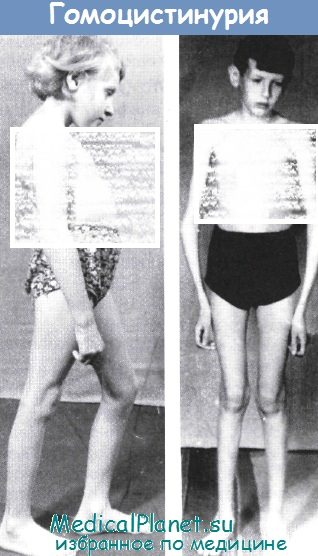
б) Гомоцистинурия. Гомоцистинурия является аутосомно-рецессивным заболеванием, вызванным мутациями с утратой функции в гене CBS, который кодирует цистатионин-β-синтетазу. Дефицит фермента вызывает накопление гомоцистеина и метионина в крови. Большинство случаев гомоцистинурии диагностируют с помощью программ скрининга новорожденных.
Клинические проявления заболевания разнообразны и включают поражение глаз (эктопия хрусталика — смещение хрусталика), центральной нервной системы (умственная отсталость, замедление физического и умственного развития, судороги, психические расстройства), скелета (марфаноподобный синдром, а также генерализованный остеопороз), сердечно-сосудистой системы (артериальные и венозные тромбозы) и кожи (гипопигментация).
Лечение включает соблюдение диеты с ограничением метионина и повышенным содержанием цистина, а также большие дозы пиридоксина.