Термин «врожденные мышечные дистрофии» относится к группе наследственных заболеваний с ранним (пренатальным, неонатальным или ранним детским) началом и гистологическими признаками, указывающими на дистрофический процесс. Он используется, чтобы охватить несколько разл. заболеваний, которые имеют общую характеристику тяжелого поражения при рождении или в раннем детстве, но которые, по иронии судьбы, часто следуют более доброкачественному клиническому течению, чем предполагают раннее начало и гистопатологические изменения в мышечной биопсии.
Отличительной чертой врожденных дистрофий, в отличие от др. мышечных дистрофий, является высокая ассоциация с пороками развития ГМ, особенно с нарушениями развития коры, такими как лиссэнцефалия/пахигирия и полимикрогирия, часто осложняющимися тяжелой эпилепсией (рис. 1). Большинство врожденных мышечных дистрофий наследуются АуР-способом.
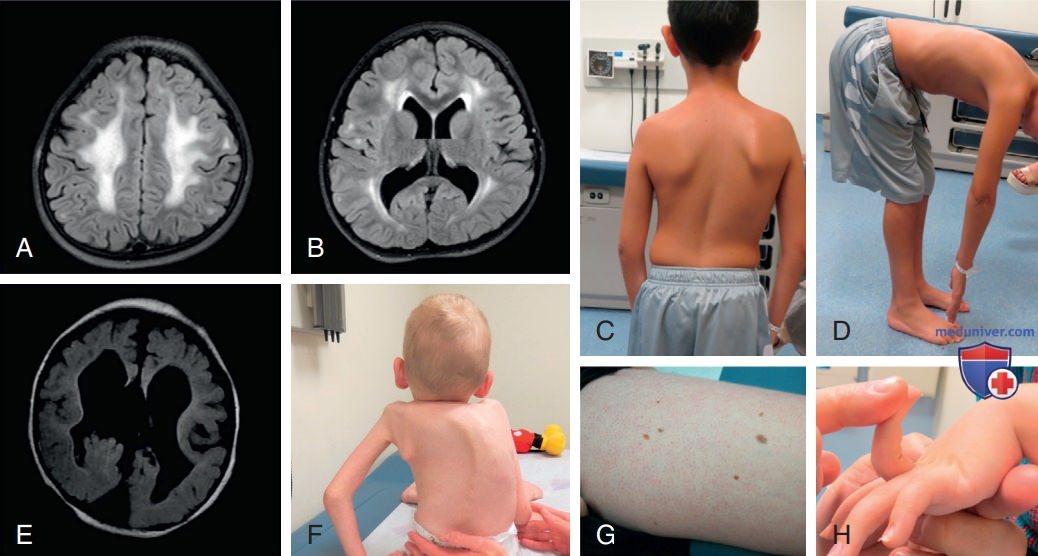
Рисунок 1. Нарушения при врожденных мышечных дистрофиях: A, B — аксиальные MPT-изображения головного мозга FLAIR, показывающие повышенную гиперинтенсивность Т2, наблюдаемую в белом в-ве у пациентов с врожденной мышечной дистрофией LAMA2; C — лопаточное крыло и сколиоз у пациента с мышечной дистрофией SEPN1; D — ригидный позвоночник, отмеченный при сгибании вперед у пациента с мышечной дистрофией SEPN1; E — МРТ головного мозга у пациента с болезнью мышца-глаз-мозг с отсутствующим мозолистым телом, мальформацией Денди-Уокера, субэпендимальными кистами; F — ранний и тяжелый кифосколиоз у ребенка с врожденной мышечной дистрофией Ульриха; G — фолликулярный кератоз, обычная кожа у больных с врожденной мышечной дистрофией, связанной с коллагеном VI; H — дистальная гипермобильность у пациента с врожденной мышечной дистрофией, связанной с коллагеном VI.
а) Клинические проявления. При нескольких разл. клинических и генетических заболеваниях, сгруппированных под общим термином «врожденные мышечные дистрофии», у младенцев часто наблюдаются контрактуры, артрогрипоз при рождении и диффузная гипотония. В некоторых случаях слабость в младенчестве может быть менее значительной, и начальные двигательное развитие даже бывает нормальным.
Мышечная масса снижена в туловище и конечностях. Контроль над положением головы часто бывает плохим из-за слабости шеи и выраженной осевой гипотонии. Лицевые мышцы могут быть слабо задействованы, но офтальмоплегия, слабость глотки и слабое сосание не являются характерными. Редко у детей выявляют дисфагию и требуется стома. Рефлексы с сухожилий могут быть снижены или отсутствовать.
Дистальный артрогрипоз характерен для всех форм врожденной мышечной дистрофии. Врожденные контрактуры, включающие аксиальные или проксимальные суставы (напр., локти), часто наводят на мысль о врожденной мышечной дистрофии Ульриха из-за мутации(мутаций) в одном из трех генов коллагена VI (COL6A1, COL6A2, COL6A3).
Врожденные мышечные дистрофии можно классифицировать по типу белка, измененного специфическими генетическими мутациями. Заболевания белков внеклеточного матрикса включают LAMA2-родственные врожденные мышечные дистрофии (дефицит мерозина, мутация LAMA2 в локусе 6q22-q23) и COL6-родственные врожденные мышечные дистрофии (врожденная мышечная дистрофия Ульриха в более тяжелой форме, миопатия Бетлема в более легкой форме заболевания) (мутации COL6A1, -А2 и -А3 в локусах 21q22 и 2q37). Белок эндоплазматического ретикулума (мутация SEPN1 в 1р35) лежит в основе синдрома ригидного позвоночника.
Аномальное гликозилирование α-дистрогликана вызывает синдром Уокера-Варбурга (мутация РОМТ1 в 9q34), мышечно-глазо-мозговую болезнь Сантавуори (мутация POMGnT1 в 1р32), мышечную дистрофию Фукуямы (мутация FCMD в 8q31-q33 и 9q31) и врожденную мышечную дистрофию с вторичным дефицитом мерозина (мутация FKRP в 19q13).
Мутации в генах, влияющих на гликозилирование а-дистрогликана, могут также приводить к более мягким или более поздним фенотипам мышечной дистрофии конечностей (с интеллектуальным вовлечением или без него). Дефекты гликозилирования (дистрогликанопатии) приводят к нарушению миграции нейробластов в ГМ плода, а также могут вызвать ДКМП. Молекула дистрогликана взаимодействует как с белками плазматической (сарколеммальной) мембраны, так и с белками внеклеточного матрикса и базальной пластинки не только в мышцах, но и в ГМ, где дефектный дистрогликан и нарушенное гликозилирование приводят к разрывам в пограничной пиальной мембране, прерывистым глиальным лимитанам, вызывающим лиссэнцефалию и глионейронную гетеротопию сверхмигрированных нервных клеток при формировании коры ГМ.
Врожденная мышечная дистрофия типа Фукуямы является второй по распространенности мышечной дистрофией в Японии (после МДД); она также зарегистрирована у детей голландского, немецкого, скандинавского и турецкого этнического происхождения. В разновидности Фукуямы тяжелая кардиомиопатия и пороки развития мозга обычно сопровождают поражение скелетных мышц. Характерны признаки и симптомы, связанные с этими органами: кардиомегалия и сердечная недостаточность, умственная отсталость, судороги, микроцефалия и неспособность к развитию.
Заболевание ЦНС может сопровождать все формы врожденной мышечной дистрофии, кроме болезни Фукуямы. Психический и неврологический статус является наиболее изменчивым признаком; по-видимому, нормальный ГМ и нормальный интеллект не исключают диагноза, если др. проявления указывают на эту миопатию. Церебральные пороки развития не последовательны и варьируют от тяжелых дисплазий (голопрозэнцефалия, лиссэнцефалия) до более легких состояний (агенезия мозолистого тела, очаговая гетеротопия коры ГМ и подкоркового белого в-ва, гипоплазия мозжечка).
Приступы являются частым осложнением уже в неонатальном периоде и могут включать инфантильные спазмы и др. тяжелые инфантильные эпилепсии.
Врожденная мышечная дистрофия является постоянной ассоциацией с церебральным дисгенезом при синдроме Уокера-Варбурга и при мышечно-глазо-мозговых заболеваниях. Патологоанатомические находки — это аномалии миграции нейробластов в коре ГМ, мозжечке и стволе ГМ. Исследования показывают значительно большее генетическое совпадение между формами врожденной мышечной дистрофии Уокера-Варбурга, Фукуямы и мышечно-глазо-мозговой болезни, которые объясняют смешанные и переходные фенотипы, так что, напр., ген, связанный с фукутином (FKRP), может вызвать миопатию Уокера-Варбурга или болезни мышца-глаз-мозг, или POMGnT1 также может продуцировать фенотипы, отличные от классической болезни Уокера-Варбурга.
б) Лабораторные результаты. Уровень креатинкиназы в сыворотке крови обычно умеренно повышен от нескольких сотен до многих тысяч МЕ/л; иногда наблюдается лишь незначительное повышение. ЭМГ определяют неспецифические миопатические особенности. Исследование всех форм врожденной мышечной дистрофии должно включать оценку сердечной деятельности и визуализацию ГМ. Биопсия мышц необходима для постановки диагноза, но если есть высокая степень подозрения болезни (напр., подтвержденный генетический дефект у родственника первой линии или четкий фенотип), то специфическое генетическое тестирование поможет избежать биопсии мышц.
в) Диагноз. Биопсия мышц является диагностической в неонатальном периоде или после него. Обширная пролиферация эндомизиального коллагена обволакивает отдельные мышечные волокна даже при рождении, заставляя их округляться в поперечном сечении, действуя как жесткий рукав, особенно во время сокращения. Перимизиальная соединительная ткань и жир также увеличиваются, и фасцикулярная организация мышцы может быть нарушена фиброзом.
Тканевые культуры внутримышечных фибробластов показывает повышенный синтез коллагена, но структура коллагена нормальная. Мышечные волокна различаются по диаметру, и многие из них имеют центральные ядра, ми-офибриллярное расщепление и др. цитоархитектурные изменения. Видны рассеянные дегенерирующие и регенерирующие волокна. Воспаления или аномальных включений не обнаружено.
Иммуноцитохим. реактивность на мерозин (α2-цепь ламинина) в сарколеммальной области отсутствует в 40% случаев и нормально выражена в остальных случаях (рис. 2 и 3). Мерозин — это апротеин, который связывает сарколеммальную мембрану миофибры с базальной пластинкой или базальной мембраной. Мерозин также экспрессируется в ГМ и в шванновских клетках. Наличие или отсутствие мерозина не всегда коррелирует с тяжестью миопатии или предсказывает ее течение. Адалин (а-дистрогликан) м.б. снижен в той или иной степени при альфа-дистрогликанопатиях, а также может наблюдаться вторичное снижение мерозина (ламинин 211).
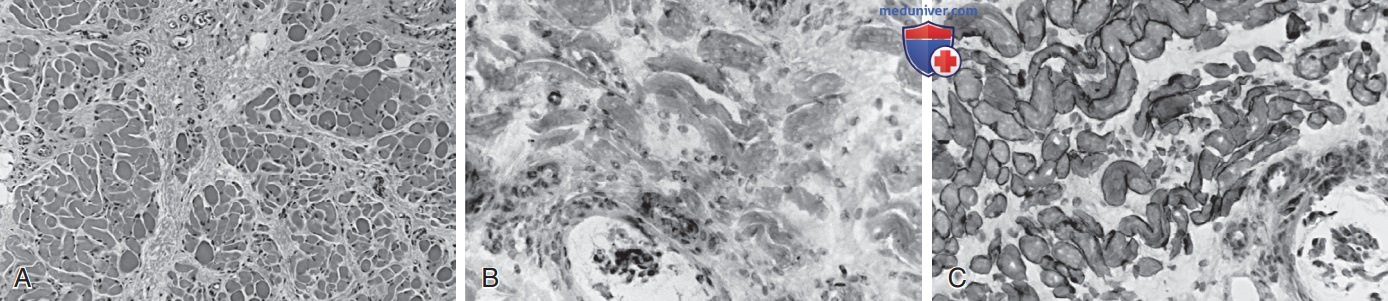
Рисунок 2. Биопсия четырехглавой мышцы бедра девочки 6 мес с врожденной мышечной дистрофией, связанной с дефицитом мерозина (а2-ламинина). Гистологически мышца инфильтрирована большим количеством коллагеновой соединительной ткани; миофибры различаются по диаметру, но некротические волокна встречаются редко (A. Иммуноцитохим. реактивность на мерозин (α2-ламинин) отсутствует во всех волокнах, включая в/фузальные миофибры мышечного веретена, видимые снизу (B). Экспрессия дистрофина (домен стержня) нормальна (C).
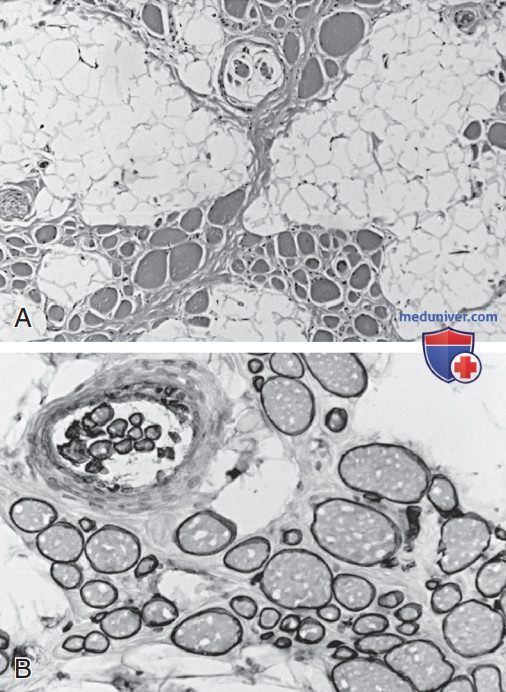
Рисунок 3. Биопсия четырехглавой мышцы бедра у девочки 2 лет с врожденной мышечной дистрофией: A — фасцикулярная архитектура мышцы сильно нарушена, и мышца заменена жиром и соединительной тканью; видны оставшиеся небольшие группы миоволокон переменного размера, включая мышечное веретено наверху; B — экспрессия мерозина нормальная как в экстрафузальных волокнах всех размеров, так и в интрафузальных веретенообразных волокнах. Тяжесть миопатии не связана с наличием или отсутствием мерозина при врожденной мышечной дистрофии. Сравните с рис. 2.
Коллаген VI избирательно редуцируется, отсутствует или неправильно локализуется в CMD, связанных с COL6. Митохондриальная дисфункция может быть еще одним вторичным дефектом.
г) Лечение. Поддерживающая терапия в это время является основой лечения. В 2010 г. опубликовано консенсусное заявление по ведению пациентов с врожденными мышечными дистрофиями (Ванг и др.). Учитывая высокую распространенность респираторной недостаточности в этой популяции, важно при каждом посещении проводить скрининг на респираторную функцию с тестированием функции легких и получением информации о частоте и продолжительности респираторных заболеваний, частоте инфекций НДП, аномальном дыхании во сне, повышенной дневной усталости или головных болях.
Исследования сна следует проводить на ранней стадии (особенно при врожденной мышечной дистрофии, связанной с коллагеном VI, и мышечной дистрофии SEPN1), когда нарушение дыхания может возникнуть даже у амбулаторных пациентов из-за прогрессирующей диафрагмальной слабости. Дополнительные респираторные поддержки могут включать в себя физиотерапию ГК, лечение кашля с отсасыванием, BiPAP и, на более продвинутых стадиях, инвазивную вентиляцию или варианты вентиляции sip/puff для непрерывной поддержки вентиляции легких.
Увеличение МТ должно быть оптимизировано, чтобы убедиться, что пациент не теряет МТ или не набирает избыточную МТ. Глотание следует оценивать на предмет выявления дисфагии. Некоторые дети будут нуждаться в питании из G-трубки из-за недостаточного перорального приема для удовлетворения потребностей в калориях, в то время как др. могут требовать почти полного питания из G-трубки из-за трудностей с глотанием. Логопедическая терапия м.б. необходима для оценки дисфагии, но также и потому, что некоторые из этих детей будут иметь некоторые трудности с артикуляцией из-за оромоторной слабости, которая может повлиять на общение в раннем возрасте. Запоры возникают часто, и их следует лечить с помощью диеты или ЛП, нормализующих стул.
Физиотерапевты должны быть вовлечены в работу с пациентами на вспомогательных устройствах, с применением растяжек связок, чтобы попытаться замедлить прогрессирование контрактур или справиться с ними. У детей может развиться сколиоз (или, при связанных с коллагеном VI врожденных мышечных дистрофий, кифосколиозные деформации), и они должны регулярно наблюдаться ортопедами для определения необходимости фиксации или хирургического вмешательства. Дети с альфа-дистрогликанопатиями с поражением ЦНС могут нуждаться в дополнительной поддержке, включая логопедию, индивидуальные образовательные программы для обучения при интеллектуальных нарушениях, лечение приступов и лечение спастичности.