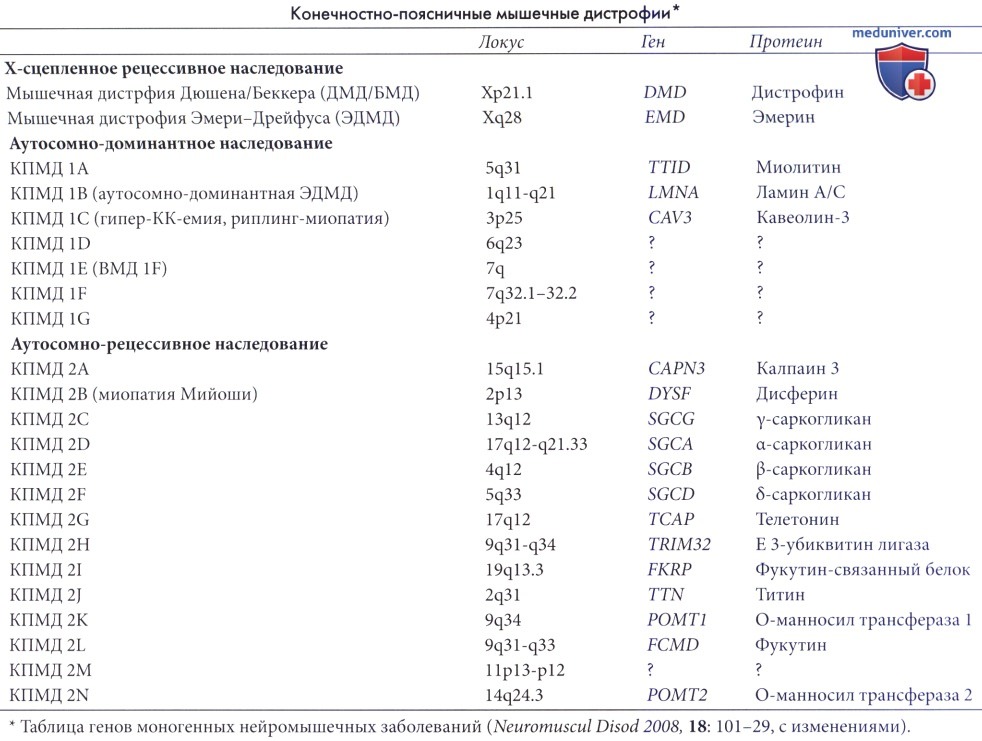
Мышечная дистрофия Эмери-Дрейфуса вызвана мутациями, включающими белки ядерной оболочки. Впервые описана как редкое Х-сцепленное рецессивное расстройство, вызванное мутациями в гене EMD, кодирующем эмерин. Обычный локус с генетической аномалией находится на длинном плече в большой области Xq28, которая включает другие мутации, вызывающие миотубулярную миопатию, неонатальную адренолейкодистрофию и тип инконтиненции пигмента Блоха-Зульцбергера; он далек от гена МДД на коротком плече Х-хромосомы.
Впоследствии обнаружено, что доминантные мутации в гене LMNA, расположенном на хромосоме 1q21, кодирующем белок ядерной оболочки lamins А/С, мутируют у пациентов мужского и женского пола. Эта форма может проявляться как врожденной мышечной дистрофией, так и с началом позже в подростковом или взрослом возрасте, и она представляет широкий фенотипический спектр. Помимо двигательных нарушений, которые изменчивы, существует также риск внезапной смерти от фибрилляции желудочков, причем в некоторых случаях может наступить уже в детском возрасте.
Клинические симптомы варьируют от врожденной мышечной дистрофии с выраженной слабостью, ДН и контрактурами с младенчества или у детей со слабостью и синдромом опущенной головы до более классических форм мышечной дистрофии Эмери-Дрейфуса. В детских формах симптомы могут начаться в 5-15 лет, но многие пациенты доживают до поздней взрослой жизни из-за медленного прогрессирования заболевания.
Мышцы не псевдогипертрофированы. Контрактуры локтевых, лодыжечных и шейных мышц-разгибателей развиваются рано, и мышцы истощаются при лопаточно-плечевом распределении. Слабость мышц лица обычно не возникает; т.о. это заболевание клинически отличается от АуД лопаточно-плечевого и лопаточно-лопаточного синдромов нейрогенного происхождения. Миотония отсутствует. Интеллектуальная функция в норме. ДКМП является тяжелой и часто является причиной смерти, чаще от дефектов проводимости, таких как ФП/трепетание предсердий и внезапная фибрилляция желудочков, чем от трудноизлечимой миокардиальной недостаточности.
Инсульт — еще одно осложнение, вторичное по отношению к сердечной аритмии. ДН более выражена при ранних и тяжелых формах и может потребовать ИВЛ, особенно у тяжелых врожденных больных. Значение креатинкиназы в сыворотке крови лишь слегка или умеренно повышено, что еще больше отличает это заболевание от др. Х-сцепленных рецессивных мышечных дистрофий.
Неспецифический некроз миофибрилл и эндомизиальный фиброз наблюдаются при биопсии мышц. Многие центронуклеарные волокна и селективная гистохимическая атрофия мышечных волокон I типа могут вызвать путаницу с миотонической дистрофией.
а) Генетика. Дефектный ген в Х-сцепленной форме называется EMD/EDMD и кодирует белок эмерин. В отличие от др. дистрофий, при которых дефектный ген экспрессируется на сарколеммальной мембране, эмерин экспрессируется на внутренней ядерной мембране; этот белок стабилизирует ядерную мембрану против механических напряжений, возникающих во время мышечного сокращения. Он взаимодействует с генами Nesprin-1 и Nesprin-2, также критически важными для целостности ядерной мембраны. Полное удаление EDMD происходит в 25% случаев и является результатом инверсии в области Xq28; полное отсутствие эмерина проявляется как вестерн-блоттингом, так и иммунореактивностью в срезах тканей.
Другой ген, LMNA в локусе 1q21, связан с ядерной оболочкой и кодирует ламины А и С, иногда называемые ламинопатией. Эта генетическая мутация вызывает сходный клинический фенотип с дефектами EMD, за исключением того, что поражаются оба пола и передается либо как АуД, либо как АуР-признак. Большинство делеций EMD являются нулевыми мутациями, в то время как >80% изменений LMNA вызваны миссенс-мутациями, причем меньшинство мутаций являются нонсенс-мутациями или мутациями вне кадра. Белок десмин также может мутировать и проявляться аномально в мышечной биопсии. Гомозиготные нонсенс-мутации в этих генах ламина А/С летальны из-за кардиомиопатии и нарушений проводимости. Есть еще много пациентов с фенотипом мышечной дистрофии Эмери-Дрейфуса, где основной генетический дефект остается неизвестным.
б) Диагностика. В подозрительных случаях дефицит эмерина может быть выявлен не только при биопсии мышц с помощью иммунореактивности и методов вестерн-блоттинга, но и во множестве др. тканей, включая циркулирующие лимфоциты периферической крови, эксфолиативные клетки слизистой оболочки щек и фибробласты кожи. Эмерин отсутствует в разл. пропорциях у женщин-носителей. Гистология мышц у пациентов с геном LMNA обычно неспецифична с миопатическими или легкими дистрофическими изменениями, с вариабельностью размера волокон, увеличением соединительной ткани и некротическими волокнами. Генетическое тестирование конкретных генов также доступно.
Пациенты должны пройти тщательное обследование сердца, включая ЭКГ, ЭхоКГ и, по крайней мере, 24-часовой холтеровский мониторинг. Следует измерять уровень креатинкиназы в сыворотке крови, поскольку он может быть умеренно повышен; хотя он и неспецифичен, это обеспечивает исходную базу для сравнения с будущими показателями. МРТ мышц ягодичных и нижних конечностей может быть полезной, особенно при мутациях гена LMNA. ЭМГ не является резюмирующим методом диагностики, но она обеспечивает последовательное наблюдение за прогрессированием миопатии. Биопсия мышц проводится с момента появления симптомов.
В ДД синдром Эмери-Дрейфуса с контрактурами суставов, легкой слабостью и более поздними сердечными симптомами обусловлен мутациями fhl1 миофибриллярной миопатии, но редуцирующие органы отсутствуют.
Лечение должно быть поддерживающим, с особым вниманием к нарушениям сердечной проводимости, и может потребоваться прием ЛС или кардиостимулятора. В настоящее время доступны имплантируемые кардиовертеры-дефибрилляторы, которые предотвращают внезапную смерть у некоторых пациентов с мышечной дистрофией Эмери-Дрейфуса. У пациентов с сердечной аритмией или выраженным снижением функции ЛЖ м.б. повышен риск тромбоэмболических осложнений, и могут потребоваться антитромботические ЛП. Легочная помощь должна включать мониторинг с помощью тестов легочной функции, а также наблюдение за нарушением дыхания во сне, если это клинически показано.
Могут быть полезными ортопедическое лечение, использование ортопедических устройств или физиотерапия, чтобы попытаться минимизировать либо замедлить скорость прогрессирования контрактур.