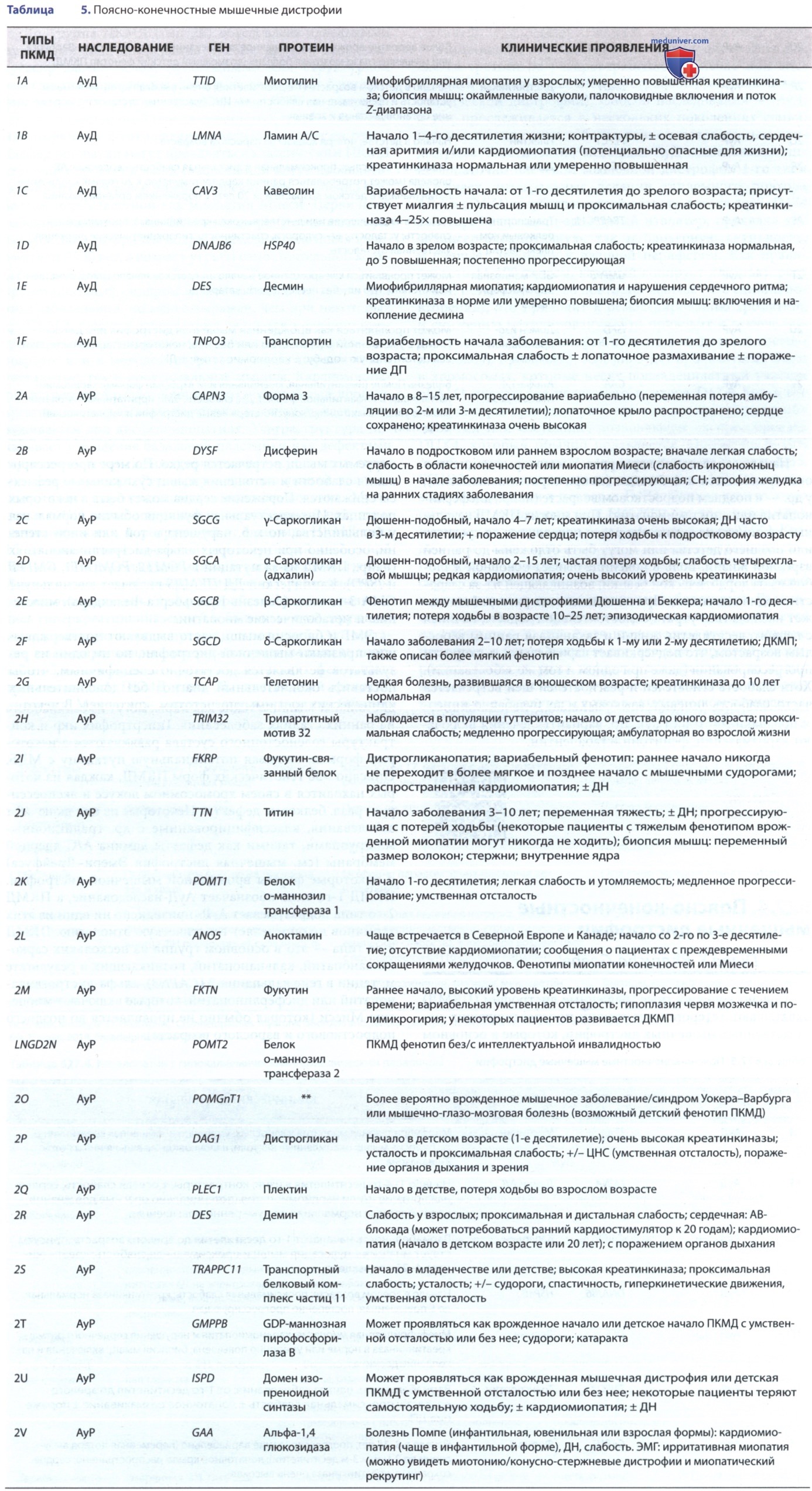
Поясно-конечностные мышечные дистрофии у детей - кратко с точки зрения педиатрии
Поясно-конечностные мышечные дистрофии (ПКМД) охватывают гетерогенную группу прогрессирующих наследственных мышечных дистрофий, которые в основном поражают мышцы тазобедренного и плечевого поясов (табл. 5). Дистальные мышцы также со временем становятся атрофичными и слабыми, и в некоторых подтипах дистальные мышцы, такие как икры, могут иметь слабость на ранних стадиях заболевания.
Гипертрофия икр и контрактуры голеностопного сустава развиваются в некоторых формах, вызывая потенциальную путаницу с МДБ. Описано >30 генетических форм ПКМД, каждая из которых находится в своем хромосомном локусе и экспрессирует разл. белковые дефекты. Некоторые из них включают заболевания, классифицированные с др. традиционными группами, такими как дефекты ламина-А/С ядерной мембраны (см. мышечная дистрофия Эмери-Дрейфуса) и некоторые формы врожденной мышечной дистрофии.
ПКМД 1-го типа обозначает АуД-наследование, а ПКМД 2-го типа подразумевает АуР- признак, но ни один из этих терминов не определяет генетическую этиологию. ПКМД 2-го типа — это в основном группа из нескольких саркогликанопатий, кальпанопатий, возникающих в результате мутации в гене кальпаина-3 (CAPN3), альфа-дистрогликанопатий или дисферлинопатий, которые включают миопатию Миеси (которая обычно не проявляется до позднего подросткового и взрослого возраста).
Начало заболевания варьирует. У некоторых пациентов оно проявляется к 4-5 годам (напр., саркогликанопатии), у др. — в позднем подростковом возрасте (напр., дисферлинопатия или аноктаминопатия). При многих ПКМД начальные клинические симптомы редко появляются до среднего или позднего детства или могут быть отложены до ранней взрослой жизни.
Боль в пояснице бывает основной жалобой из-за лордотической осанки, возникающей из-за слабости ягодичных мышц. При многих из этих расстройств может наблюдаться утрата самостоятельной ходьбы, начиная с первого десятилетия жизни и заканчивая ранним взрослым возрастом, что подчеркивает вариабельность скорости прогрессирования (даже при одном и том же заболевании). Хотя слабость сгибателей и разгибателей шеи встречается часто, слабость лицевых, языковых и др. бульбарно-иннервируемых мышц встречается редко.
По мере прогрессирования слабости и истощения мышц сухожильные рефлексы снижаются. Поражение сердца может быть в некоторых подтипах. Интеллектуальная функция обычно нормальная у большинства, но м.б. нарушена в той или иной степени, особенно при некоторых альфа-дистрогликанопатиях (напр., ПКМД из-за мутаций в РОМТ2, POMGnTl, GMPPB и ISPD). Клинический ДД ПКМД включает ювенильную/СМА 3-го типа (болезнь Кугельберга-Веландера), миастению и метаболические миопатии.
ЭМГ и биопсия мышц часто выявляют подтверждающие признаки мышечной дистрофии, но ни один из результатов не является достаточно специфичным, чтобы поставить окончательный диагноз без дополнительных клинических или иммуногистохим. критериев. В некоторых случаях α-саркогликан (ранее известный как адалин), связанный с дистрофином гликопротеин сарколеммы, является дефицитным; этот специфический дефект может быть обнаружен при биопсии мышц с помощью иммуноцитохимии, как и дефицит трех др. форм саркогликана. Повышенный уровень креатинкиназы в сыворотке крови типичен, но уровень его повышения варьируется в зависимости от семьи. ЭКГ обычно остается неизменной.
Мутантный дистрофин-ассоциированный белок в саркогликановом комплексе (саркогликанопатия; ПКМД типов 2С, 2Е и 2F) отвечает за некоторые случаи АуР-формы ПКМД. Большинство саркогликанопатий являются результатом мутации в α-саркогликане; др. ПКМД, также встречаются возникающие в результате дефицита β-, γ- и δ-саркогликана.
В нормальной гладкой мускулатуре α-саркогликан заменяется ε-саркогликаном, а остальные остаются такими же. Дистрогликанопатии вызываются мутациями, приводящими к аномальному гликозилированию альфа-дистрогликана, и независимо от гена все мутации, по-видимому, влияют на функцию дистрогликана. Гистохимически дистрогликанопатии часто имеют дефекты (потерю или снижение) иммунореактивности к одному из двух АТл: VIA41 или IIH6, которые распознают углеводные фрагменты Альфа-дистрогликана. Степень снижения может варьироваться от едва заметного до сильного.
Др. группа ПКМД (тип 2В) обусловлена аллельными мутациями гена дисферлина (DYSF), другого гена, экспрессирующего белок, необходимый для структурной целостности сарколеммы, хотя и не связанный с дистрофин-гликопротеиновым комплексом. DYSF взаимодействует с кавеолином-3 или кальпаином-3, и дефицит DYSF м.б. вторичным по отношению к дефектам в этих др. генах. Дисферлинопатии могут проявляться классическим ПКМД паттерном проксимальной слабости или могут проявляться ранней слабостью в нижних конечностях, особенно слабостью икр, известной как миопатия Миеси.
Первичный дефект кальпаина-3 (тип 2А) имеет широкую клиническую вариабельность, причем возраст начала заболевания колеблется от 2-40 лет, а возраст утраты самостоятельной ходьбы — от 5 лет до конца 30-х годов. Респираторный компрометационный синдром может наблюдаться позже при этом заболевании, но менее выражен, чем при некоторых др. ПКМД. Оба эти заболевания являются медленно прогрессирующими миопатиями с началом в подростковом возрасте или в молодом возрасте и могут поражать как дистальные, так и проксимальные мышцы. Кардиомиопатия встречается редко. Хронически повышенный уровень креатинкиназы в сыворотке крови в тысячах случаев обнаруживается при дисферлинопатиях.
Ультраструктура показывает утолщение базальной пластинки над дефектами сарколеммы и замещение сарколеммы несколькими слоями мелких везикул. Регенерирующие мышечные волокна превосходят вырождающиеся мышечные волокна. Мутации в CAV3 также могут иметь вариабельный нейромышечный фенотип, варьирующий от фенотипа ПКМД типа 1С до дистальной миопатии, пульсирующей мышечной болезни, гиперкреатинкиназемии и непереносимости ФН. Имеются также сообщения о пациентах с рабдомиолизом с кавеолинопатиями. Эти расстройства раньше назывались гиперкреатинкиназемией и болезнью пульсирующих мышц, которую иногда путают с миотонией.
АуР-мутация в активированном кальцием хлоридном канале аноктамин-5 может вызывать один из следующих фенотипов: проксимальный ПКМД типа 2L; дистальный фенотип миопатии Миеси; или гиперкреатинкиназемию.
Обычно она проявляется во взрослом возрасте и чаще встречается в Северной Европе и Канаде. По-видимому, никакой сопутствующей кардиомиопатии нет, хотя есть сообщения о ранних преждевременных сокращениях желудочков.
Существует генетическое перекрытие группы ПКМД с врожденными мышечными дистрофиями, такими как синдром Уокера-Варбурга с РОМТ, мышечная дистрофия Фукуямы с генетическими дефектами FKRP и GMPPB. Пациенты с мутациями в этих генах могут иметь ранний CMD-подобный фенотип к детскому или более позднему фенотипу ПКМД, и в обоих моторных фенотипах могут иметь или не иметь разл. степень умственной отсталости.