Лице-лопаточно-плечевая мышечная дистрофия является третьей по распространенности мышечной дистрофией (после мышечной дистрофии Дюшенна и миотонической дистрофии). Способ наследования — АуД, и ген прослеживается в нескольких поколениях семьи, причем последующее в более раннем возрасте, чем предыдущее. Генетический механизм АуД-наследования лице-лопаточно-плечевой мышечной дистрофии 1-го типа включает интегральные делеции тандемного повтора 3,3 КБ (D4Z4) в субтеломерной области локуса 4q35.
D4Z4 действует как ламин-зависимый изолятор, проявляя как энхансер-блокирующую, так и барьерную активность, и смещает теломеру к ядерной периферии. Как правило, есть 11-100 тандем, который копирует или повторяет D4Z4. Когда количество повторов меньше (<10 единиц повторения), это приводит к ремоделированию хроматина и снижению метилирования, что приводит к включению экспрессии DUX4 (которая обычно находится в состоянии покоя).
Кроме того, болезнь будет проявляться только в хромосомах, которые несут полиаденилатный участок pLAM1 дистальнее последнего повтора D4Z4. Когда все эти факторы присутствуют, это создает «разрешающий» гаплотип или состояние, позволяющее экспрессировать DUX4, который обычно подавляется. Примерно 5-10% семей с этим фенотипом не соответствуют локусу 4q35. FSHD2, хотя клинически перекрывается с FSHD1, не вызван сокращением в повторах D4Z4.
Однако вместо этого он вызван мутациями гена SMCHD1 (на хромосоме 18р), которые могут привести к гипометилированию D4Z4. Когда эти мутации существуют в условиях «разрешающего» гаплотипа и сигнала полиаденилирования, DUX4 экспрессируется, снова разделяя конечный общий путь, ведущий к одному и тому же клиническому заболеванию. Распространенность варьируется географически, но колеблется от 1:8000 до 1:20 000. Хотя клиническое начало обычно наступает в более позднем детстве или взрослой жизни, ранние молекулярные дефекты, возникающие во время миогенеза, проявляются у человеческого плода, и заболевание может развиваться уже в младенческом периоде.
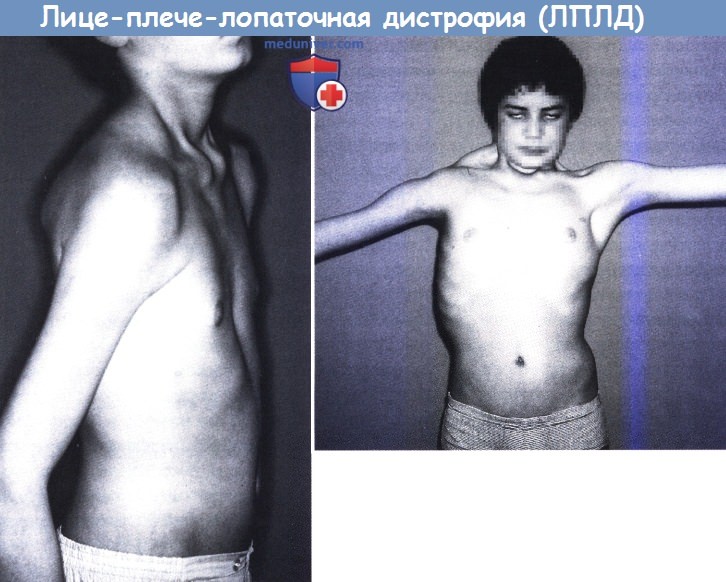
а) Клинические проявления. При лице-лопаточно-плечевой дистрофии наблюдается самая ранняя и тяжелая слабость мышц лица и плечевого пояса. Асимметричная или неравномерная слабость, когда она присутствует, должна вызывать подозрение на лице-лопаточно-плечевую дистрофию. Слабость лицевых мышц отличается от миотонической дистрофии; вместо перевернутой V-образной верхней губы рот при лице-лопаточно-плечевой дистрофии округлый и кажется сморщенным, потому что губы выступают.
Неспособность полностью закрыть глаза во сне является распространенным выражением слабости верхней части лица; у некоторых пациентов наблюдается экстраокулярная мышечная слабость, хотя офтальмоплегия редко бывает полной. В редких случаях с синдромом Мебиуса была связана лице-лопаточно-плечевая дистрофия. Слабость глотки и языка может отсутствовать и никогда не бывает такой сильной, как мышц лица. Потеря слуха, которая может быть субклинической, и васкулопатия сетчатки (неотличимая от болезни Коутса) являются сопутствующими признаками, особенно в тяжелых случаях лице-лопаточно-плечевой дистрофии с ранним детским началом.
Крыловидные лопатки проявляются и часто даже у младенцев. Наблюдается уплощение или даже вогнутость дельтовидного контура, а бицепсы и трицепсы плечевых мышц истощены и слабы.
Лице-плече-лопаточная дистрофия у 13-летнего мальчика. Выраженное поражение плечевого пояса, больше справа.
Невозможность полностью закрыть глаза (вверху). Тот же пациент (слева), отмечается атрофия мышц шеи, амиотрофия мышц плеча и крыловидная лопатка.
Мышцы тазового пояса и бедер со временем теряют силу и подвергаются атрофии, появляются симптом Говерса и походка Тренделенбурга. Контрактуры конечностей встречаются редко. Слабость пальцев и запястий иногда является первым симптомом. Слабость передней большеберцовой и малоберцовой мышц может привести к падению стопы; это осложнение обычно возникает только в запущенных случаях с выраженной слабостью. Поясничный лордоз и кифосколиоз являются распространенными осложнениями поражения осевых мышц. Псевдогипертрофия голеней не является обычным признаком, описывается редко.
Существует большая клиническая вариабельность, в т.ч. в семьях. Лице-лопаточно-плечевая мышечная дистрофия может быть легким заболеванием, вызывающим минимальную инвалидизацию. Клинические проявления могут не наблюдаться в детском возрасте и становятся заметными к середине взрослой жизни. В более тяжелых случаях пациенты м.б. в раннем возрасте. Ок. 20% пациентов теряют самостоятельную ходьбу, а ок. 10-15% пациентов могут нуждаться в неинвазивной или инвазивной респираторной поддержке. В отличие от большинства других мышечных дистрофий, асимметрия слабости является характерной особенностью.
У 30% пациентов заболевание протекает бессимптомно или проявляется только в виде легкого нарушения движения лопатки и снижения сухожильных рефлексов, о которых они не знали до проведения обычного неврологического обследования.
б) Лабораторные результаты. Сывороточные уровни креатинкиназы и др. ферментов сильно варьируют, от нормальных или почти нормальных до увеличения в несколько тысяч. ЭКГ должна быть выполнена, несмотря на то что результат, как правило, нормальный. ЭМГ выявляет неспецифические миопатические потенциалы мышц. Диагностическое молекулярное тестирование в отдельных случаях и в семьях показано для прогнозирования.
в) Диагностика и дифференциальная диагностика. Молекулярно-генетическая диагностика является наиболее специфичным подтверждением, если клинические подозрения высоки, с семейным анамнезом заболевания или без него. Мышечная биопсия различает более одной формы лице-лопаточно-плечевой дистрофии, что согласуется с клиническими данными о том, что несколько разл. заболеваний охватываются термином «лице-лопаточно-плечевая дистрофия». Мышечная биопсия и ЭМГ также отличают первичную миопатию от нейрогенного заболевания с аналогичным распределением мышечного поражения.
Общие гистопатологические находки в мышечном биопсийном материале включают обширную пролиферацию соединительной ткани между мышечными волокнами, экстремальное изменение размера волокон со многими гипертрофическими, а также атрофическими миофибрами и рассеянными дегенерирующими и регенерирующими волокнами. Выделяют также воспалительный тип лице-лопаточно-плечевой мышечной дистрофии, характеризующийся обширными лимфоцитарными инфильтратами внутри мышечных пучков. Несмотря на сходство этой формы с воспалительными миопатиями, такими как полимиозит, нет никаких признаков аутоиммунного заболевания, а стероиды и иммуносупрессивные ЛП не изменяют клинического течения.
Точный гистопатологический диагноз имеет важное терапевтическое значение. Воспаление мононуклеарных клеток в образце биопсии мышц у детей <2 лет обычно является лице-лопаточно-плечевой дистрофией или, реже, врожденной мышечной дистрофией.
г) Лечение. Следует регулярно следить за функцией легких, и если есть опасения по поводу дневных головных болей или повышенной усталости, следует провести исследование сна для оценки любого нарушения дыхания во сне или апноэ во сне. Легкие аэробные упражнения и режимы растяжки могут помочь предотвратить декондиционирование или атрофию с течением времени. Высокоинтенсивные тренировки и силовые тренировки или поднятие тяжестей не рекомендуются, потому что они не помогут восстановить силу либо замедлить прогрессирование слабости или истощения мышц.
Стопы и сколиоз можно лечить ортопедическими средствами. В отдельных случаях хирургическая коррекция лопаток (операция фиксации лопатки) к грудной стенке обеспечивает стабильность плеча и отведение руки, но плечевая плексопатия, синдром «замороженного плеча» и переломы лопатки возможны в виде осложнений. Дополнительные варианты реабилитации для лопаточной поддержки включают кинезиотейпирование. Хроническая боль обычно наблюдается у пациентов с лице-лопаточно-плечевой дистрофией и может потребовать дальнейшего лечения, включая габапентин, ТЦА или ФН и КПТ.
Косметическое улучшение мимических мышц лица может быть достигнуто реконструктивной хирургией, которая прививает фасцию lata к скуловой мышце и к скуловой головке мышцы quadratus labii superioris. Упражнение на лицевые мышцы может помочь свести к минимуму вторичную атрофию неиспользования. Следует проводить рутинные обследования глаз (тестирование на болезнь Коутса), а у маленьких больных детей — аудиограммы. В настоящее время клинически не существует эффективного фармакологического или генетического лечения.