Миотоническая мышечная дистрофия — вторая по распространенности мышечная дистрофия в Северной Америке, Европе и Австралии, частота которой в общей популяции 1:20 000-1:100 000. Она наследуется как АуД-признак. Классическая миотоническая дистрофия 1 типа (болезнь Штейнерта) вызывается экспансией тринуклеотида CTG на хромосоме 19q13.3 в 3’-нетранслируемой области DMPK, гена, кодирующего серин-треонин протеинкиназу.
Миотоническая дистрофия 2-го типа связана с нестабильной экспансией тетрануклеотидного повтора CCTG на хромосоме 3q21 интрона гена белка цинкового пальца 9. Третья поздняя форма миотоническая дистрофии идентифицируется в локусе 15q21-q24.
Миотоническая дистрофия является примером генетического дефекта, вызывающего дисфункцию в разл. системах органов. Поражается не только поперечнополосатая мускулатура, но и гладкие мышцы ЖКТ и матки, изменяется сердечная функция, у пациентов возникают множественные и вариабельные эндокринопатии, иммунологические нарушения, катаракта, дисморфизм лица, повышается риск ЗНО, умственных нарушений и др. неврологических отклонений.
а) Клинические проявления. Миотоническая дистрофия 1-го типа становится симптоматическим в любом возрасте, но миотоническая дистрофия 2-го типа редко проявляется в младенчестве или раннем детстве. При обычном клиническом течении, исключая тяжелую неонатальную форму, дети с миотонической дистрофией 1-го типа могут казаться почти нормальными при рождении, или же истончение мышц лица и гипотония уже м.б. ранними проявлениями заболевания.
Характерен внешний вид лица, состоящий из перевернутой V-образной верхней губы, тонких щек и зубчатых вогнутых височных мышц (рис. 1). Голова может быть узкой, а нёбо высоким и изогнутым, потому что слабые височные и крыловидные мышцы в конце жизни плода не оказывают достаточного бокового воздействия на развивающуюся голову и лицо.
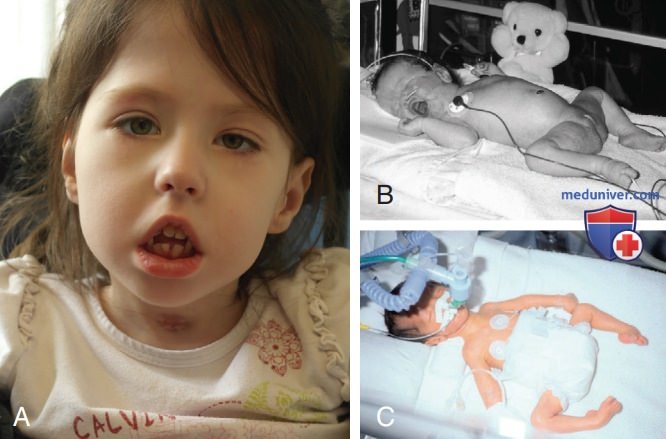
Рисунок 1. 6-летний ребенок с врожденной миотонической дистрофией с 1975 цитозин-тимин-гуаниновыми повторами в гене DMPK, характерное удлиненное лицо, левосторонний птоз и открытый, повернутый вниз (тентованный) рот с неправильным прикусом зубов: А — рубец от трахеостомы свидетельствует о тяжелом респираторном дистрессе, требующем интубации во время рождения; B — новорожденный с врожденной миотонической дистрофией также с открытым, повернутым вниз ртом и лягушачьим положением нижних конечностей; C — новорожденный с врожденной миотонической дистрофией с РДС и артрогрипозом.
При миотонической дистрофии 1-го типа мышечная слабость часто бывает легкой в первые несколько лет (детская форма) или может даже не проявляться до подросткового возраста либо ранней зрелости (классическая/взрослая форма). Прогрессирующее истончение дистальных мышц становится все более очевидным, особенно в мышцах рук. Тенарные и гипотенарные возвышения уплощены, атрофия оставляет глубокие борозды между пальцами. Дорсальные мышцы предплечий и мышцы переднего отдела голеней также истощаются.
Язык тонкий, атрофичный. Истончение грудино-ключично-сосцевидных мышц придает шее длинный тонкий цилиндрический контур. Проксимальные мышцы также в конце концов подвергаются атрофии, и появляется лопаточное колебание. Трудности с подъемом по лестнице и симптом Говерса прогрессируют. Сухожильные рефлексы обычно сохраняются.
Дистальное распределение мышечного истончения при миотонической дистрофии является исключением из общего правила миопатий. Мышечная атрофия и слабость при миотонической дистрофии медленно прогрессируют в детском и подростковом возрасте и продолжаются в зрелом возрасте. Пациенты с миотонической дистрофией редко теряют способность ходить даже в конце взрослой жизни, хотя для стабилизации лодыжек могут потребоваться шины или фиксаторы.
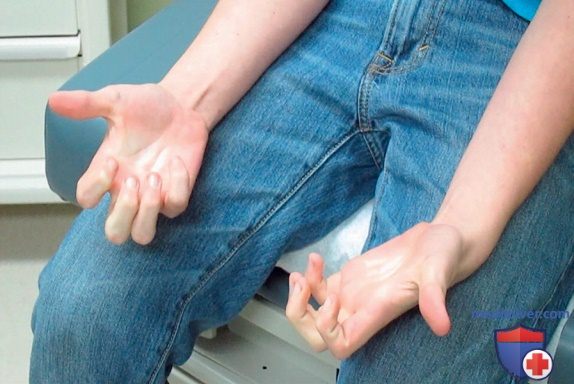
Миотония — общая характерная черта для нескольких др. миопатий, не возникает в младенчестве и обычно не проявляется клинически или даже на ЭМГ до ~5 лет. Миотония — это очень медленное расслабление мышц после сокращения, независимо от того, было это сокращение добровольным или вызвано рефлексом растяжения либо электрической стимуляцией. При физикальном осмотре миотонию можно продемонстрировать, попросив пациента сжать кулаки, а затем быстро разжать руки (миотония захвата; рис. 2).
Рисунок 2. Пациента попросили сжать обе руки в течение нескольких секунд, а затем внезапно отпустить хватку, и прошло несколько секунд, прежде чем было достигнуто полное расслабление, что было обнаружено при обследовании, известном как миотония захвата.
Она может быть вызвана ударом по тенору резиновым молоточком (перкуссионная миотония), и ее можно обнаружить, наблюдая за непроизвольным движением большого пальца по ладони. Миотонию также можно увидеть на языке, прижав край деревянной лопатки к его тыльной поверхности и наблюдая глубокую борозду, которая медленно исчезает. Тяжесть миотонии необязательно совпадает со степенью слабости, и самые слабые мышцы часто имеют лишь минимальную миотонию. Миотония — это не болезненный мышечный спазм. Скелетно-мышечные боли и усталость довольно часто наблюдаются у пациентов с миотонической дистрофией.
Речь больных миотонической дистрофией часто плохо артикулирована и невнятная из-за поражения мышц лица, языка и глотки. Как миотония, так и мышечная слабость могут вызывать трудности в речи и глотании пациентов. Иногда возникают трудности с глотанием, пациенты могут подвергаться риску аспирационной пневмонии. Неполная внешняя офтальмоплегия иногда является следствием слабости экстраокулярных мышц.
Поражение гладкой мускулатуры ЖКТ приводит к медленному опорожнению желудка, плохой перистальтике и запорам. У некоторых пациентов энкопрез связан со слабостью анального сфинктера. Женщины с миотонической дистрофией могут иметь неэффективные или измененные сокращения матки во время родов.
Поражение сердца обычно проявляется как сердечная блокада в проводящей системе Пуркинье и аритмия (и внезапная смерть), а не как кардиомиопатия, в отличие от большинства др. мышечных дистрофий. Предсердные или желудочковые тахиаритмии приводят к внезапной смерти у взрослых и детей старшего возраста.
Эндокринные нарушения затрагивают многие железы и проявляются в любое время в течение болезни, поэтому эндокринный статус должен ежегодно пересматриваться. Гипотиреоз встречается часто; гипертиреоз встречается редко. Недостаточность коры надпочечников может привести к аддисонову кризу даже в младенчестве. СД часто встречается у пациентов с миотонической дистрофией; у некоторых детей наблюдается нарушение высвобождения инсулина, а не его дефектная выработка.
Наступление половой зрелости может быть преждевременным или, чаще, запоздалым. Атрофия яичек и дефицит тестостерона нередко встречаются у взрослых и являются причиной высокой частоты мужского бесплодия. Атрофия яичников встречается редко. Лобное облысение характерно для пациентов мужского пола и часто начинается в подростковом возрасте.
При миотонической дистрофии часто встречаются иммунологические нарушения. Уровень IgG в плазме крови нередко бывает низким.
Катаракта часто возникает при миотонической дистрофии. Они м.б. врожденными или начаться в любое время в детском или взрослом возрасте. Ранние катаракты выявляются только при осмотре щелевой лампой; рекомендуется периодическое обследование у офтальмолога. Зрительные вызванные потенциалы часто нарушены у детей с миотонической дистрофией и не связаны с катарактой. Они обычно не сопровождаются нарушением зрения.
Около половины пациентов с миотонической дистрофией умственно отсталые, но тяжелые нарушения нехарактерны. Остальные имеют средний или иногда выше среднего интеллект. Эпилепсия встречается редко. Когнитивные нарушения могут быть результатом накопления мутантной мРНК DMPK и аберрантного альтернативного сплайсинга в нейронах коры ГМ. Более высокая, чем ожидалось, частота РАС встречается у детей с миотонической дистрофией 1-го типа.
Тяжелая врожденная форма миотонической дистрофии появляется у меньшинства детей, рожденных от матерей с симптоматической миотонической дистрофией (см. рис. 1). Все пациенты с этим тяжелым врожденным заболеванием на сегодняшний день имели форму DM1. Симптомы могут проявляться пренатально с многоводием и уменьшением движений плода.
При рождении пациенты, как правило, имеют выраженную гипотонию, трудности с дыханием или ДН и трудности с кормлением, и у них м.б. дополнительные ортопедические заболевания, такие как косолапость или более обширные врожденные контрактуры (артрогрипоз multiplex congenita).
Заметно истончение мышц лица. Младенцам может потребоваться искусственное вскармливание или ИВЛ при слабости дыхательных мышц либо апноэ. Нуждающиеся в вентиляции <30 дней часто выживают. При длительной вентиляции уровень детской смертности 25% и более низкая вероятность выживания без вентиляции. Дети, вентилируемые до 30 дней, имеют лучшие двигательные, языковые и повседневные навыки, чем те, которые требуют длительной вентиляции. Одна или обе створки диафрагмы могут быть нефункциональными.
Живот становится раздутым газом в желудке и кишечнике из-за плохой перистальтики и слабости гладкой мускулатуры. Вздутие живота еще больше затрудняет дыхание. Невозможность опорожнить прямую кишку может усугубить проблему. Миотония обычно не отсутствует при врожденной форме в течение неонатального периода, но может быть в детском возрасте (обычно после 5 лет).
б) Лабораторные результаты. Классическая миотоническая электромиограмма не встречается у младенцев, но может появиться у детей в раннем школьном возрасте. Уровни сывороточной креатинкиназы и др. сывороточных ферментов в мышцах м.б. нормальными или только слегка повышенными в сотни (никогда в тысячи) раз.
ЭКГ следует проводить ежегодно в раннем детском возрасте. УЗИ БП м.б. показано больным детям для определения функции диафрагмы. Могут потребоваться РОГК и БП, а также дополнительные исследования моторики ЖКТ или исследования глотания.
Эндокринная оценка должна проводиться для определения функции ЩЖ и коры надпочечников, а также для проверки углеводного обмена (тест на толерантность к глюкозе). Следует исследовать Ig и, при необходимости, проводить более обширные иммунологические исследования.
в) Диагноз. Первичным диагностическим тестом является анализ ДНК крови, чтобы выявить аномальное увеличение повтора CTG или CCTG. Возможна и пренатальная диагностика. Биопсия мышц у детей старшего возраста показывает много мышечных волокон с центральными ядрами и селективную атрофию гистохимических волокон I типа, но дегенерирующие волокна обычно немногочисленны и широко разбросаны, и фиброз мышц практически отсутствует. Интрафузальные волокна мышечных веретен также изменены.
У маленьких детей с распространенной формой заболевания образец биопсии может даже казаться нормальным или, по крайней мере, не выявлять некрозов миофибры, что является поразительным контрастом с МДД. При тяжелой неонатальной форме миотонической дистрофии биопсия мышц выявляет задержку созревания на разл. стадиях развития у одних и врожденную диспропорцию типа мышечных волокон у других. Вполне вероятно, что сарколеммальная мембрана мышечных волокон не только обладает патологическими свойствами электрической поляризации, но и не способна реагировать на трофические воздействия двигательного нейрона.
Мышечная биопсия обычно не требуется для постановки диагноза, который в типичных случаях может быть основан на клинических симптомах, включая семейный анамнез. Неонатальную миотоническую дистрофию, вызывающую множественный артрогрипоз и/или тяжелую неонатальную гипотонию, следует отличать от амиоплазии, врожденной мышечной дистрофии с экспрессией мерозина или без нее, врожденной миастении, СМА и артрогрипоза, вторичного по отношению к олигогидрамниозу.
г) Генетика. Генетический дефект миотонической мышечной дистрофии находится на хромосоме 19 в локусе 19q13. Он состоит из экспансии гена DM, кодирующего серин-треонинкиназу (DMPK), с многочисленными повторами кодона CTG. Экспансии варьируют от 50 до >2000, при этом нормальные аллели этого гена варьируют по размеру от 5-37; чем больше экспансия, тем тяжелее клиническая экспрессия, причем наибольшие экспансии наблюдаются при тяжелой неонатальной форме.
Редко болезнь ассоциируется с отсутствием обнаруживаемых повторов, возможно, спонтанной коррекцией предыдущего увеличения, но феноменом, все еще не до конца понятым. Др. миотоническая дистрофия (проксимальная миотоническая миопатия) — клиническое образование, связанное по крайней мере с двумя другими хромосомными локусами, чем классическая миотоническая дистрофия, но с одним локусом, который разделяет общий уникальный патогенез, опосредованный мутантной мРНК. Дефекты сплайсинга РНК объясняют инсулинорезистентность при миотонических дистрофиях, а также миотонию.
Клиническая и генетическая экспрессия может варьировать между братьями и сестрами или между больным родителем и ребенком. При тяжелой неонатальной форме заболевания мать является передающим родителем в 94% случаев, но у мужчин не характеризуется только повышенным мужским бесплодием. Сообщалось о нескольких случаях передачи заболевания от отца. Генетический анализ показывает, что новорожденные с симптоматической формой обычно имеют гораздо больше повторов кодона CTG, чем пациенты с более классической формой заболевания, независимо от того, какой родитель болен.
Миотоническая дистрофия часто имеет паттерн, в котором каждое последующее поколение имеет тенденцию заболеть чаще, чем предыдущее. Существует пренатальная генетическая диагностика миотонической дистрофии.
д) Лечение. Специфического лечения не существует, но сердечные, эндокринные, желудочно-кишечные и глазные осложнения часто поддаются лечению. Физиотерапевтическое и ортопедическое лечение контрактур при неонатальной форме заболевания м.б. полезным. Миотония может улучшиться при ФН (феномен разминки), и следует избегать экстремальных холодных температур. Следует проводить кардиологическое наблюдение с ежегодной ЭКГ, а также холтеровские исследования и ЭхоКГ примерно каждые 2 года.
Имплантация кардиостимулятора может быть рассмотрена при блокаде сердца, и антиаритмические ЛП м.б. показаны, но они необходимы только в редких случаях у детей. Необходимо решить респираторные проблемы; стратегии лечения могут включать BiPAP, помощь при кашле и стимулирующую спирометрию.
Миотония может быть уменьшена, и функция восстановлена ЛС, которые повышают порог деполяризации мышечных мембран, такими как мексилетин, фенитоин, карбамазепин, прокаинамид и хинидин сульфат. Эти ЛП также обладают кардиотропным действием, поэтому перед их назначением важно оценить состояние сердца. Фенитоин и карбамазепин используются в дозах, аналогичных тем, которые используются в качестве противоэпилептических ЛС; следует поддерживать концентрацию в сыворотке крови 10-20 мкг/мл для фенитоина и 5-12 мкг/мл для карбамазепина. Если инвалидность пациента вызвана главным образом мышечной слабостью, а не миотонией, то ЛС не будут иметь никакой ценности.
Избыточная сонливость иногда контролируется с помощью метилфенидата или модафинила. Низкие или умеренные ФН м.б. полезны при миалгиях.
Меры предосторожности необходимы при анестезии. Следует учитывать более высокую частоту осложнений при анестезии у пациентов с миотонией. Сукцинилхолина следует избегать из-за риска миотонии, и вместо этого следует использовать недеполяризующие миорелаксанты короткого действия, дозы которых зависят от степени истощения мышц. Следует использовать модифицированную индукцию быстрой последовательности для интубации. Во время выздоровления неостигмин следует применять с осторожностью, а экстубация должна происходить, когда пациент более полно просыпается. После седации пациенты должны находиться под пристальным наблюдением из-за риска аспирации.
е) Другие миотонические синдромы. Большинство пациентов с миотонией имеют миотоническую дистрофию. Однако миотония не специфична для этого заболевания и встречается в нескольких более редких состояниях.
Миотоническая хондродистрофия (болезнь Шварца-Ямпеля) — редкое врожденное заболевание, характеризующееся генерализованной мышечной гипертрофией и слабостью. Дисморфические фенотипические особенности и рентгенографический вид длинных костей напоминают болезнь Моркио, но аномальных мукополисахаридов не обнаружено. Наблюдается карликовость, патология суставов, блефарофимоз. У некоторых пациентов установлено кровное родство, что свидетельствует об АуР-типе наследования. Мышечный белок перлекан, кодируемый геном SJS1, большим гепарансульфатным протеогликаном базальных мембран и хряща, является дефектным в некоторых случаях болезни Шварца-Ямпеля и объясняет как мышечную гипервозбудимость, так и хондродисплазию.
ЭМГ выявляет непрерывную электрическую активность в мышечных волокнах, близкую или идентичную миотонии. Биопсия мышц выявляет неспецифические миопатические признаки, которые в одних случаях минимальны, а в др. выражены. Саркотубулярная система расширена.
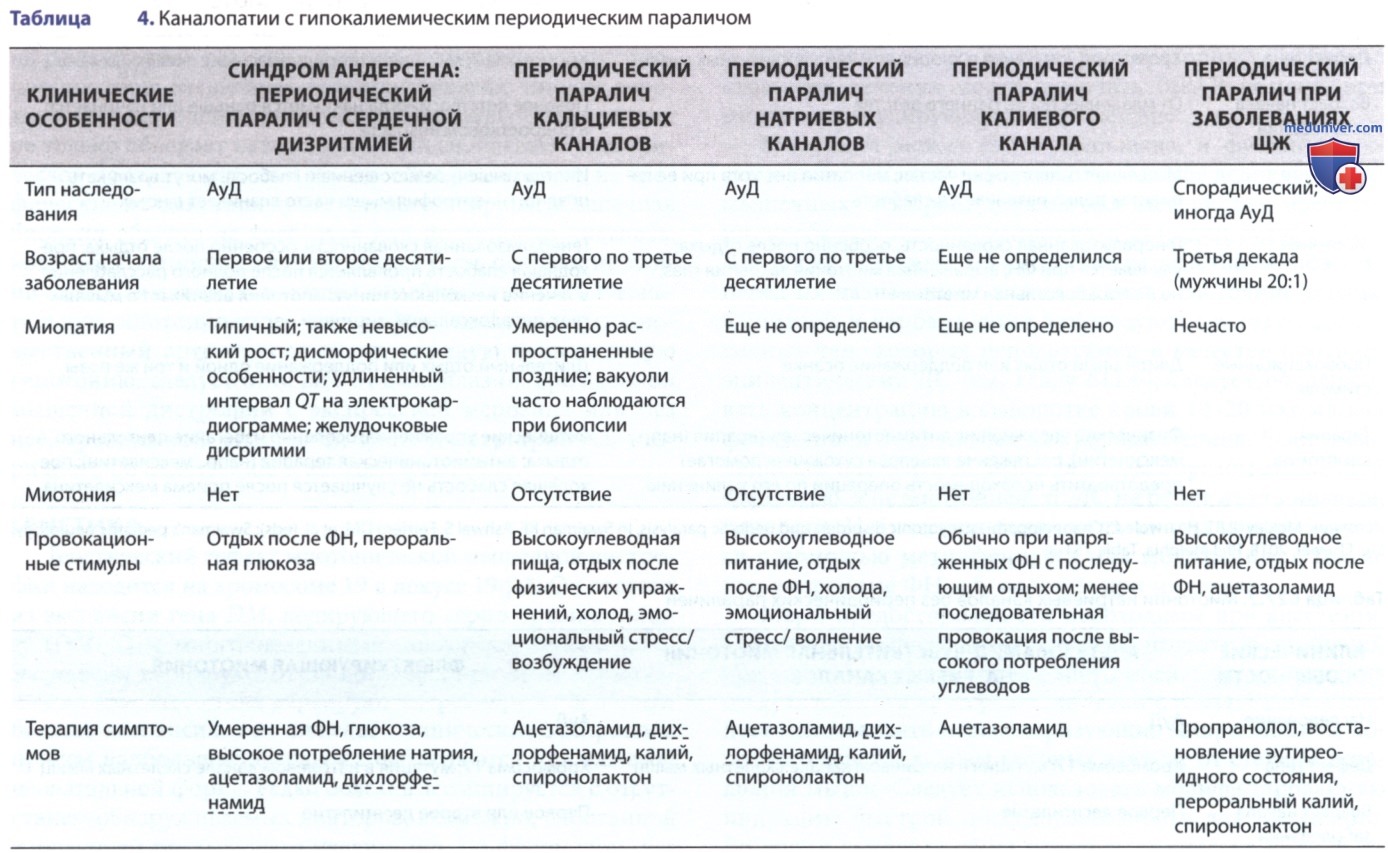
Врожденная миотония (болезнь Томсена), тип каналопатии, является наиболее распространенным из синдромов недистрофической миотонии (табл. 1-4) и характеризуется слабостью и генерализованной мышечной гипертрофией, так что пораженные дети напоминают культуристов («внешность Геркулеса»). Миотония выражена и может развиться в возрасте 2-3 лет, раньше, чем при миотонической дистрофии. Болезнь клинически стабильна и, по-видимому, не прогрессирует в течение многих лет. Биоптаты мышц показывают минимальные патологические изменения, а ЭМГ демонстрирует миотонию. Разл. семьи описываются с АуД (болезнь Томсена) либо АуР (болезнь Беккера, не путать с МДБ или МДД) наследованием. Мутации могут быть нонсенс, миссенс, или знаковыми.
Однако, в частности, миссенс-мутации, которые изменяют активацию димера CLC-1, приводят к доминантным наследственным формам заболевания. Пациенты с АуР-формой (болезнь Беккера), как правило, имеют более тяжелое заболевание. АуД и АуР-формы врожденной миотонии сопоставлены с одним и тем же локусом 7q35. Этот ген важен для целостности хлоридных каналов сарколеммальной и Т-канальцевой мембран.
Парамиотония — связанная с температурой миотония, которая усугубляется холодом и смягчается теплыми внешними температурами. Пациенты испытывают трудности при купании в холодной воде или если они одеты неадекватно в холодную погоду. Парамиотония — врожденная (болезнь Эйленбурга), дефект гена в локусе 17q13.1-13.3, идентичном локусу, идентифицированному при гиперкалиемическом периодическом параличе. В отличие от врожденной миотонии, парамиотония — расстройство натриевого канала, вызванное мутацией в α-субъединице. Миотоническая дистрофия является натриевой каналопатией (см. табл. 3).
При натриевых каналопатиях физические упражнения вызывают усиление миотонии, тогда как при хлоридных каналопатиях физические упражнения уменьшают миотонию. Это легко проверить во время обследования, попросив пациентов закрыть глаза с усилием и открыть их несколько раз; это становится все труднее при расстройствах натриевых каналов и все легче при расстройствах хлоридных каналов.
Лечение недистрофических миотоний включает мексилетин в качестве первой линии (как для миотоний натриевых каналов, так и для миотоний хлоридных каналов). Показано, что мексилетин уменьшает неподвижность и признаки миотонии рук. Др. варианты лечения включают карбамазепин, фенитоин и габапентин.