P.S. Клинические рекомендации по тематике статьи на момент ее подготоки отсутствуют.
Мышечная дистрофия отличается от всех др. нервно-мышечных заболеваний четырьмя обязательными критериями: это первичная миопатия, имеющая генетическую основу, течение прогрессирующее, а дегенерация и гибель мышечных волокон происходят на определенной стадии заболевания. Это определение исключает нейрогенные заболевания, такие как СМА, ненаследственные миопатии, дерматомиозит, непрогрессивные и некротизирующие врожденные миопатии, врожденная диспропорция мышечных волокон и непрогрессивные наследственные метаболические миопатии.
Некоторые метаболические миопатии могут соответствовать определению прогрессирующей мышечной дистрофии, но традиционно не классифицируются как дистрофии (дефицит мышечного карнитина).
Многие мышечные дистрофии м.б. в конечном итоге переклассифицированы в метаболические миопатии, как только биохим. дефекты будут лучше определены. Мышечные дистрофии — группа неродственных заболеваний, каждое из которых передается по разным генетическим признакам и отличается своим клиническим течением и выраженностью.
Идентифицируемые мутации в некоторых генах могут привести к спектру клинических фенотипов, варьирующих по возрасту начала заболевания, тяжести и наличию сопутствующих заболеваний. Некоторые мышечные дистрофии более тяжелые, могут выявляться при рождении или вскоре после рождения, обычно определяемые как врожденные мышечные дистрофии, в то время как др. могут иметь начало в детстве или даже во взрослом возрасте.
Существует диапазон тяжести от тех, которые приводят к смерти в неонатальном периоде, до тех, которые прогрессируют постепенно в течение десятилетий, как правило, с нормальной продолжительностью жизни. Некоторые категории дистрофий, такие как поясно-конечностная мышечная дистрофия (ПКМД), являются не однородными заболеваниями, а скорее синдромами, охватывающими несколько разл. клинических образований и ряд предполагаемых генов.
Мышечная дистрофия Дюшенна (МДД) — наиболее распространенное наследственное нервно-мышечное заболевание, поражающее все расы и этнические группы. Его характерными клиническими признаками являются прогрессирующая слабость, интеллектуальные нарушения и гипертрофия икр с пролиферацией соединительной ткани и прогрессирующим фиброзом мышц. Заболеваемость среди живорожденных мальчиков составляет 1:3600. Это заболевание наследуется как Х-сцепленный рецессивный признак.
Аномальный ген находится в локусе Хр21 и является одним из самых крупных генов. Мышечная дистрофия Беккера (МДБ) — заболевание, которое в основе похоже на МДД, с генетическим дефектом в том же локусе, но клинически оно протекает более мягко и с затяжным течением.
а) Клинические проявления. МДД у мальчиков редко проявляются симптомами при рождении или в раннем младенчестве, хотя у некоторых из них может быть гипотония. Ранние грубые двигательные навыки, такие как переворачивание, сидение и стояние, обычно достигаются в соответствующем возрасте или с некоторой задержкой. Отличительные черты лица не являются ранним признаком, потому что слабость лицевых мышц появляется позже; в более позднем детстве может наблюдаться поперечная или горизонтальная улыбка.
Функция ходьбы развивается к 12 мес, но слабость тазобедренного пояса может проявляться в легкой форме уже на 2-м году. Малыши могут принимать лордотическую позу, когда стоят, чтобы компенсировать слабость ягодиц. Ранний признак Говерса можно увидеть в возрасте 3 лет, но почти всегда он проявляется в возрасте 5 или 6 лет. Тренделенбургская (ковыляющая) походка часто появляется уже в это время. Распространенные проявления у детей раннего возраста включают задержку ходьбы, частое падение, ходьбу на носках и проблемы с бегом или ходьбой по лестнице, задержку развития и, реже, злокачественную гипертермию после анестезии.
Продолжительность пребывания пациента с МДД в компенсированном состоянии варьирует. У пациентов может наблюдаться трудность с передвижением из-за слабости проксимального отдела нижних конечностей, а также усугубляться прогрессирующими контрактурами голеностопного сустава и ходьбой на носках. Возраст полной потери передвижения обычно ~10—14 лет. Возраст, в котором происходит потеря самостоятельной ходьбы, со временем увеличился с появлением клинических рекомендаций по ведению с применением ГКС (напр., преднизолона или дефлазакорта) у мальчиков с МДД (см. раздел о лечении ниже). С помощью ортопедической фиксации, физиотерапии, а иногда и незначительной хирургии (удлинение ахиллова сухожилия) большинство из них способны ходить до 12 лет.
Поддержание ходьбы важно не только для сохранения автономии в повседневной жизни (что имеет психосоциальные преимущества для пациента и его семьи), но и обеспечивает дополнительные преимущества в замедлении прогрессирования сколиоза (обычно ухудшается после потери ходьбы) и в поддержании здоровья легких.
Неуклонное прогрессирование слабости продолжается и во втором десятилетии. Функция дистальных мышц обычно достаточно хорошо сохраняется, что позволяет ребенку продолжать пользоваться столовыми приборами, карандашом и компьютерной клавиатурой. По мере прогрессирования заболевания в подростковом возрасте сила верхних конечностей еще больше снижается, и у пациентов могут прогрессировать трудности с самостоятельным поднесением рук ко рту, усталость от письма и нарастание контрактур, в т.ч. в кистях и пальцах. Поражение дыхательной мускулатуры часто проявляется слабым и неэффективным кашлем, легочными инфекциями и снижением дыхательного резерва.
Ранние легочные симптомы часто включают храп и апноэ во сне. Родители или пациенты могут сообщать о частых головных болях, затруднении пробуждения по утрам и повышенной дневной усталости как о признаках нарушения дыхания во сне. Слабость мышц глотки может привести к эпизодам аспирации, носовой регургитации жидкостей и воздушному или носовому качеству голоса. Функция экстраокулярных мышц хорошо сохраняется. Недержание мочи и кала из-за слабости анального и уретрального сфинктеров — редкое и очень позднее явление.
Контрактуры чаще всего затрагивают лодыжки, колени, бедра и локти. По мере прогрессирования слабости верхних конечностей контрактуры наблюдаются также при боковом вращении шеи, плеч и пальцев. Сколиоз часто встречается у пациентов с МДД. Деформация ГК еще больше снижает легочную емкость и сдавливает сердце. Это также может привести к большему дискомфорту и, если он достаточно серьезен, риску вывиха бедра. Сколиоз обычно прогрессирует быстрее после того, как ребенок перестает ходить.
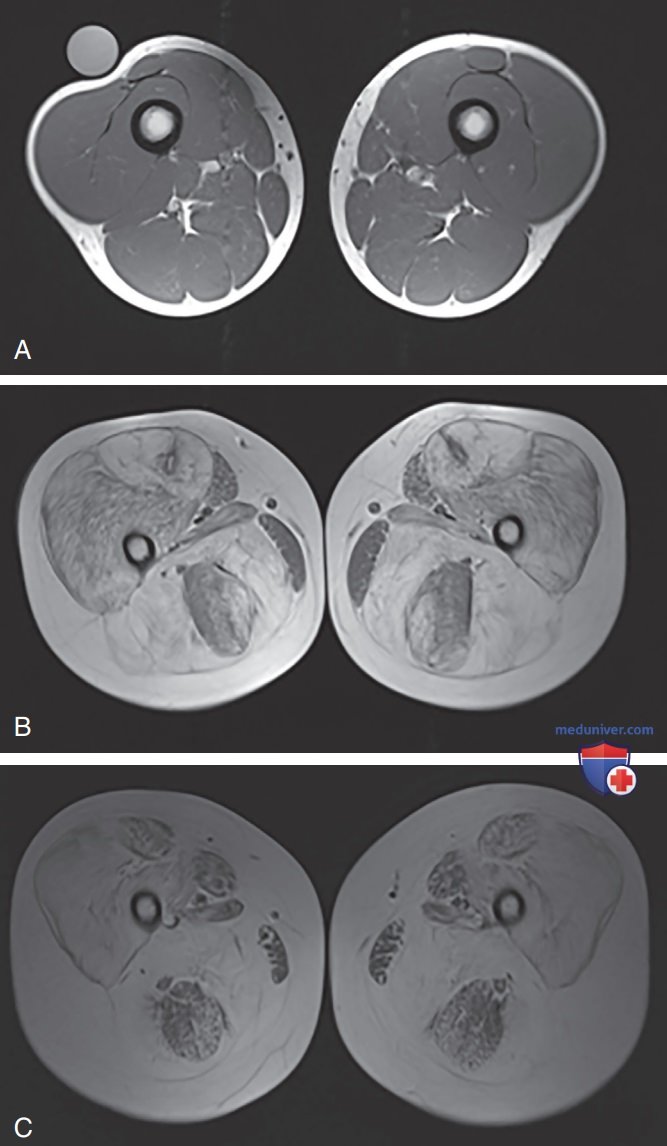
Однако в эпоху применения ГКС возможно дальнейшее защитное воздействие на развитие и скорость прогрессирования сколиоза. Увеличение икр (псевдогипертрофия) и истощение мышц бедер являются классическими признаками. Увеличение вызвано гипертрофией некоторых мышечных волокон, инфильтрацией мышц жиром и пролиферацией коллагена. После икроножных мышц следующим наиболее распространенным местом мышечной гипертрофии является язык, за которым следуют мышцы предплечья. Аномалии мышц выявляются с помощью МРТ мышц для оценки сигнала, содержания воды, фракций жира и даже профилей MP-спектроскопии (рис. 1). Фасцикуляции языка не происходит. Мышцы сфинктеров вовлекаются редко.
Рисунок 1. Осевые Т1-МРТ-изображения бедра здорового 14-летнего мальчика (А), 9-летнего (В) и 14-летнего амбулаторных мальчиков с мышечной дистрофией Дюшенна (С), показывающие повышенную жировую инфильтрацию и атрофию мышц
Если контрактуры голеностопного сустава не являются серьезными, глубокие сухожильные рефлексы лодыжки хорошо сохраняются до терминальных стадий. Сухожильные коленные рефлексы могут вызываться до ~6 лет, но они менее выражены, чем ахилловы, и в конечном итоге истощаются с прогрессированием слабости.
В верхних конечностях брахиорадиальный рефлекс обычно сильнее, чем рефлексы с бицепса или трицепса.
Кардиомиопатия, включая персистирующую тахикардию, фиброз миокарда, встречается у большинства пациентов с МДД. Тяжесть поражения сердца не обязательно коррелирует со степенью слабости скелетных мышц. У пациентов с МДД прогрессирование кардиомиопатии, как правило, происходит после утраты самостоятельного передвижения. Однако у пациентов с МДБ может развиться прогрессирование кардиомиопатии и даже тяжелая СН, несмотря на то что они все еще находятся в амбулаторных условиях. Дисфункция гладкой мускулатуры, особенно ЖКТ, является незначительной, но частой особенностью, упускаемой из виду.
Интеллектуальные нарушения встречаются у большинства пациентов, хотя только 20-30% имеют IQ<70. Степень умственной отсталости различная, некоторые пациенты нуждаются в специальном образовании и испытывают трудности с чтением и письмом, а те, кто страдает менее серьезно, могут нуждаться лишь в дополнительном обучении. Степень тяжести умственной отсталости, по-видимому, не коррелирует с тяжестью миопатии, но может быть связана с расположением мутаций в гене дистрофина. Эпилепсия встречается несколько чаще, чем в общей педиатрической популяции, хотя и не является характерной чертой МДД.
У некоторых пациентов может развиться аутистическое поведение. Дистрофин экспрессируется в ГМ и сетчатке, а также в поперечнополосатой и сердечной мышцах, хотя в ГМ его уровень ниже, чем в мышцах. Это распределение может объяснить некоторые симптомы ЦНС. Аномалии кортикальной архитектуры и дендритной арборизации м.б. обнаружены патологоанатомически; церебральная атрофия проявляется на МРТ на поздних стадиях клинического течения. Пациенты с МДД и МДБ могут иногда сообщать о миалгиях, которые часто вызваны ФН. Кальциноз мышц встречается редко.
Смерть у мальчиков с МДД наступает в позднем подростковом возрасте, до 20 лет. Причинами смерти являются ДН во время сна, трудноизлечимая СН, пневмония или, иногда, аспирация и ОДН.
При МДБ начало заболевания часто наступает после 5-7 лет, и мальчики остаются самостоятельно передвигающимися до позднего подросткового или даже зрелого возраста. Псевдогипертрофия икр, кардиомиопатия и повышенный уровень креатинкиназы в сыворотке крови аналогичны таковым у пациентов с МДД. Учитывая повышенный уровень активности у пациентов с МДБ, сердечные проявления, включая тахикардию, одышку или усталость, могут проявляться раньше у пациентов с МДБ и даже в условиях независимой амбулаторной терапии.
Неспособность к обучению встречается реже. Начало слабости наступает позже при МДБ, чем при МДД. Продолжительность жизни пациентов с МДБ обычно составляет 40-50 лет, причем имеющиеся сердечные и легочные осложнения часто обостряются.
б) Лабораторные результаты. Уровень креатинкиназы в сыворотке крови постоянно значительно повышается при МДД, даже на предсимптомных стадиях, в т.ч. при рождении. Обычная концентрация в сыворотке крови составляет 15 000-35 000 МЕ/л (нормальная <160 МЕ/л). Нормальный уровень креатинкиназы в сыворотке крови несовместим с диагнозом МДД, хотя на терминальных стадиях заболевания значение креатинкиназы в сыворотке крови м.б. значительно ниже, чем несколько лет назад, потому что остается меньше мышц для дегенерации.
Др. лизосомальные ферменты, присутствующие в мышцах, такие как альдолаза и ACT, также повышены, но менее специфичны.
Оценка состояния сердца с помощью ЭхоКГ и ЭКГ имеет важное значение и должна контролироваться. Рекомендуется проводить кардиологическое наблюдение каждые два года, начиная с момента постановки диагноза, а затем ежегодно, когда у ребенка возникают сердечные проблемы или он достигает возраста 10 лет. После установления диагноза МДД пациенты должны быть направлены к детскому кардиологу, который знаком с уходом за пациентами с МДД, для длительного кардиологического наблюдения.
МРТ сердца может обнаружить такие изменения, как фиброз мышц в сердце, который проявляется даже раньше, чем изменения, наблюдаемые при ЭхоКГ.
ЭМГ выявляет характерные миопатические признаки, но не является специфичной для МДД. Никаких признаков денервации не обнаруживается. Скорость моторной и сенсорной нервной проводимости в норме.
в) Диагноз. Генетическая оценка МДД обычно начинается с анализа делеции/дупликации гена дистрофина с использованием анализа дозировки. Если результат «-», то производится секвенирование гена дистрофина методом секвенирования следующего поколения.
Если генетический анализ на мутацию в гене дистрофина «-», но подозрение основано на клинических признаках и уровне креатинкиназы в сыворотке крови, то биопсия мышц с иммуногистохимией дистрофина может быть информативной. Иммуногистохим. окрашивание замороженных участков мышечной биоптатной ткани выявляет различия в домене стержня, карбоксильном конце (который прикрепляется к сарколемме) и N-конце или амино-конце (который прикрепляется к актиновым миофиламентам) большой молекулы дистрофина и м.б. прогностическим для клинического течения болезни Дюшенна или Беккера.
Более выраженная слабость возникает при усечении молекулы дистрофина на карбоксильном конце, чем на амино-конце. Дистрогликаны и др. регионарные белки сарколеммы, такие как мерозин и саркогликаны, также м.б. измерены, поскольку они могут быть вторично снижены. Можно было бы провести дальнейшее генетическое тестирование, которое может включать секвенирование РНК из мышц, чтобы попытаться идентифицировать мутацию, изменяющую сплайсинг (напр., ту, которая не м.б. идентифицирована при секвенировании следующего поколения).
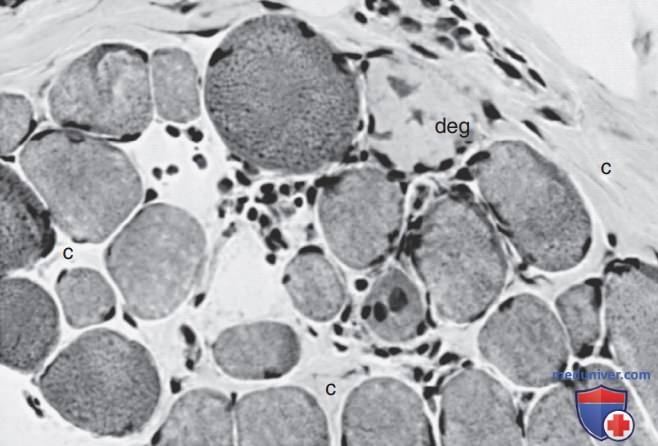
Биопсия мышц является диагностической и показывает характерные изменения (рис. 2 и 3). Миопатические изменения включают эндомизиальнуто пролиферацию соединительной ткани, рассеянные дегенерирующие и регенерирующие миофибры, очаги инфильтратов моно-нуклеарных воспалительных клеток в ответ на некроз мышечных волокон, слабые архитектурные изменения в еще функционирующих мышечных волокнах и многие плотные волокна. Эти гиперконтрактированные волокна, вероятно, являются результатом сегментарного некроза на др. уровне, позволяя кальцию проникать в место разрушения сарколеммальной мембраны и вызывать сокращение всей длины мышечного волокна. Кальцификация в миофибрах коррелирует с вторичным дефицитом β-дистрогликанов.
Рисунок 2. Биопсия мышц 4-летнего мальчика с мышечной дистрофией Дюшенна. Видны как атрофические, так и гипертрофические мышечные волокна, а некоторые волокна дегенерируют (deg). Соединительная ткань (с) между мышечными волокнами увеличена (гематоксилин и эозин, х400)
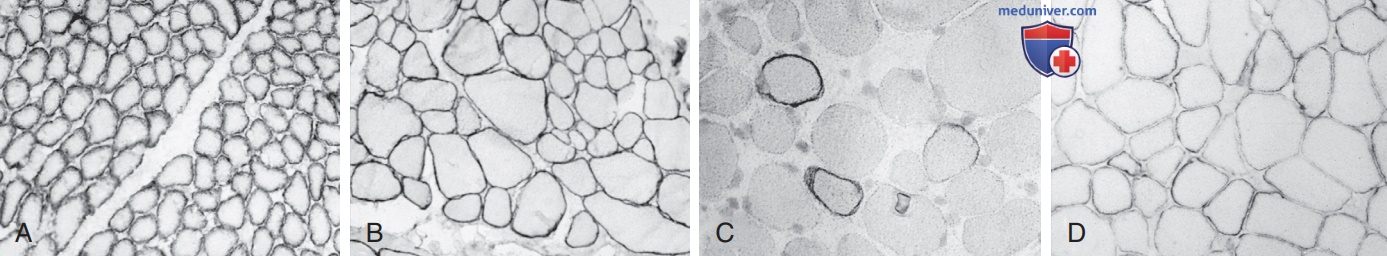
Рисунок 3. Дистрофин демонстрируется иммуногистохим. реактивностью в биоптатах мышц новорожденного мальчика в норме (A), 10-летнего мальчика с мышечной дистрофией конечностей (B), 6-летнего мальчика с мышечной дистрофией Дюшенна (C) и 10-летнего мальчика с мышечной дистрофией Беккера (D). В нормальном состоянии, а также при несвязанных мышечных дистрофиях, при которых дистрофин не поражается, сарколеммальная мембрана каждого волокна сильно окрашена, включая атрофические и гипертрофические волокна. При дистрофии Дюшенна большинство миофибр не экспрессируют обнаруживаемого дистрофина, но несколько рассеянных волокон, известных как ревер-тантные волокна, проявляют почти нормальную иммунореактивность. При мышечной дистрофии Беккера аномальная молекула дистрофина тонкая, с бледным окрашиванием сарколеммы, при котором реактивность изменяется не только между миофибрами, но и по окружности отдельных волокон (х250)
Решение о том, следует ли проводить биопсию мышц для установления диагноза, иногда представляет проблемы. При наличии семейного анамнеза заболевания, особенно в случае больных брата/сестры, диагноз которых был подтвержден, пациенту с типичными клиническими признаками МДД и высокими концентрациями креатин-киназы в сыворотке крови, вероятно, не нужно проходить биопсию. Результат генетического тестирования (анализ делеции/дупликации дистрофина и секвенирование) также может повлиять на необходимость проведения биопсии мышц. Первый случай в семье, даже если клинические признаки типичны, должен иметь подтвержденный диагноз, чтобы убедиться, что др. миопатия не маскируется под МДД. Наиболее распространенными мышцами являются vastus lateralis и gastrocnemius.
г) Генетическая этиология и патогенез. Несмотря на Х-сцепленное рецессивное наследование при МДД, ~30% случаев являются новыми или de novo мутациями, и мать не является носителем. У матери-носителя обычно отсутствует мышечная слабость, но ок. 8% матерей-носителей имеют незначительную мышечную слабость. Эти симптомы у женщин объясняются гипотезой Лайона, согласно которой нормальная Х-хромосома становится инактивированной, а та, в которой происходит делеция гена, — активной. Полная клиническая картина МДД наблюдалась у нескольких женщин с синдромом Тернера, у которых единственная Х-хромосома должна была иметь делецию гена Хр21.
Бессимптомное носительство МДД ассоциировано с повышенными значениями креатинкиназы в сыворотке крови в 50% случаев. Уровень увеличения обычно составляет сотни или несколько тысяч, но не имеет экстремальных значений, отмеченных у заболевших мужчин. Женщины в препубертатном периоде, являющиеся носителями дистрофии, также имеют повышенные значения креатинкиназы в сыворотке крови, причем самые высокие уровни наблюдаются в возрасте 8-12 лет.
Если мать заболевшего мальчика имеет нормальный уровень креатинкиназы, то маловероятно, что ее дочь м.б. идентифицирована как носитель, но нужно измерить креатинкиназу. Мышечная биопсия предполагаемых женщин-носителей может выявить дополнительно 10% тех, у кого уровень креатинкиназы в сыворотке крови не повышен; специфический генетический диагноз с использованием ПЦР в периферической крови является окончательным.
Примерно 40% женщин-носителей могут быть подвержены риску развития кардиомиопатии или фиброза (как это было показано на кардиограммах женщин-носителей), даже при отсутствии слабости скелетных мышц.
Цитоскелетный белок 427 кДА, известный как дистрофин, кодируется геном в локусе Хр21.2. Этот ген содержит 79 экзонов кодирующей последовательности и 2,5 Мб ДНК. Этот субсарколеммальный белок прикрепляется к сарколеммальной мембране, перекрывающей полосы А и М миофибрилл, и состоит из четырех разл. областей или доменов: аминоконцерн содержит 250 аминокислот и связан с N-актиновым сайтом связывания а-актинина; второй домен является самым большим, с 2800 аминокислотами, и содержит много повторов, придавая ему характерную форму стержня; третий, богатый цистеином домен связан с карбоксильным концом α-актинина; и последний карбоксильный концевой домен из 400 аминокислот уникален для дистрофина и связанного с дистрофином белка, кодируемого хромосомой 6.
Отсутствие дистрофина в сарколемме нарушает мембранный цитоскелет и приводит к вторичной потере других компонентов цитоскелета.
Молекулярные дефекты при дистрофинопатиях варьируют и включают в/генные делеции, дупликации или точечные мутации нуклеотидов. У 65% пациентов наблюдаются делеции, у 10% — дупликации и у 10% — точечные мутации или более мелкие перестройки. Менее чем в 1% случаев глубокая интронная мутация может привести к изменению сплайсинга и тем самым повлиять на рамку считывания. Место или размер в/генной аномалии не всегда хорошо коррелирует с фенотипической тяжестью; как у форм Дюшенна, так и Беккера мутации происходят в основном около середины гена, включая делеции в областях между экзонами 45-55.
Фенотипические или клинические вариации объясняются изменением трансляционной рамки считывания мессенджерной РНК (мРНК), что приводит к нестабильным, усеченным молекулам дистрофина и тяжелому, классическому МДД; мутации, которые сохраняют рамку считывания, еще позволяют переводить кодирующие последовательности дальше вниз по гену и производить полуфункциональный дистрофин, выраженный клинически как МДБ. Еще более легкая форма заболевания, ранее известная как миопатия четырехглавой мышцы, также вызвана аномальной молекулой дистрофина. Клинический спектр дистрофинопатий включает не только классические формы МДД и МДБ, но и колеблется от тяжелой неонатальной мышечной дистрофии до бессимптомных детей с персистирующим повышением уровня креатинкиназы в сыворотке крови >1000 МЕ/л.
Анализ белка дистрофина требует биопсии мышц и определяется вестерн-блоттингом или в срезах тканей иммуногистохим. методами с использованием либо флуоресцентной, либо световой микроскопии антидистрофиновой антисыворотки (см. рис. 3). При классическом МДД обнаруживаются уровни <3% нормы; при МДД молекулярная масса дистрофина снижается до 20-90% нормы у 80% пациентов, у 15% пациентов дистрофин имеет нормальный размер, но уменьшен в количестве, а у 5% пациентов имеется аномально большой белок, вызванный чрезмерными дубликациями или повторами кодонов. Селективная иммунореактивность разл. участков молекулы дистрофина в срезах мышечного биоптата различает формы МДД и МДБ (рис. 4).
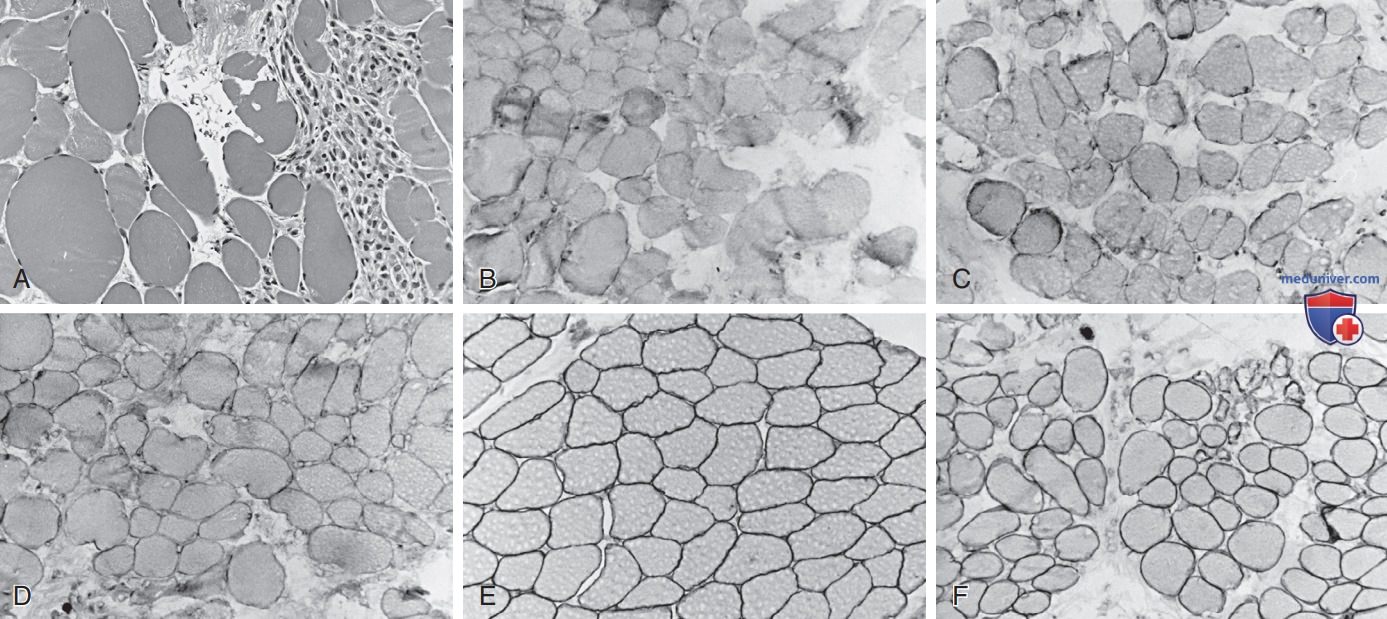
Рисунок 4. Биопсия четырехглавой мышцы бедра у 4-летнего мальчика с мышечной дистрофией Беккера (A). Миофибры сильно различаются по размеру как при атрофических, так и при гипертрофических формах; справа находится зона дегенерации и некроза, инфильтрированная макрофагами, аналогичная мышечной дистрофии Дюшенна (гематоксилин и эозин, х250). Иммунореактивность с использованием антител против молекулы дистрофина в стержневом домене (B), карбоксилтермине (C) и аминотермине (D) показывает недостаточную, но не полностью отсутствующую экспрессию дистрофина; большинство волокон всех размеров сохраняют некоторое количество дистрофина в отдельных частях сарколеммы, но не по всей окружности в поперечном сечении. В качестве альтернативы выраженность дистрофина меньше и выглядит слабой по сравнению с одновременно инкубированным нормальным контролем у др. ребенка аналогичного возраста (E). Экспрессия мерозина нормальна у этого пациента с мышечной дистрофией Беккера, как в больших, так и в малых миофибрах, и отсутствует только в откровенно некротических волокнах (F). Сравните с классической мышечной дистрофией Дюшенна, показанной на рис. 3.
Демонстрация делеций и дупликаций также может быть сделана из образцов крови с помощью более быстрой ПЦР, которая идентифицирует до 98% делеций путем амплификации 18 экзонов, но не может обнаружить дупликации. Т.о., диагноз м.б. подтвержден на молекулярно-генетическом уровне либо из мышечного биоптата, либо из периферической крови, хотя до 30% мужчин с МДД или МДБ имеют ложнонормальную ПЦР крови; все случаи дистрофинопатии выявляются при биопсии мышц.
Те же методы анализа ДНК из образцов крови могут быть применены для выявления носителей у родственников женского пола, находящихся в группе риска, таких как родные и двоюродные сестры, и для определения того, является мать носителем или произошла новая мутация в эмбрионе. Пренатальная диагностика возможна уже на 12-й неделе беременности путем отбора проб ворсинок хориона для анализа ДНК методом саутерн-блотт или ПЦР, а в случаях абортированных плодов с МДД мышца имеет патологическое окрашивание дистрофином с помощью иммуногистохимии.
д) Лечение. В настоящее время нет никакого мед. лечения этой болезни. До сих пор основным методом лечения МДД была поддерживающая и профилактическая помощь. Многое можно сделать для лечения осложнений и улучшения качества жизни пострадавших детей.
Показано, что ГКС (преднизон или дефлазакорт) замедляют снижение мышечной силы и увеличивают время, в течение которого пациент может самостоятельно ходить, и это может иметь дополнительные преимущества при прогрессировании сколиоза. Начало приема стероидов показано, когда у ребенка наблюдается плато в развитии и/или регресс в двигательном развитии по сравнению со сверстниками. Обычно это происходит к 4-6 годам. Рекомендуемые дозы — преднизон 0,75 мг/кг/сут или дефлазакорт 0,9 мг/кг/сут. Альтернативные протоколы введения ГКС включают дозирование только в выходные дни, альтернативные дневные режимы или 10-дневные режимы включения/выключения; ежедневный режим предпочтительнее, что подтверждено на сравнительных исследованиях.
Долгосрочные осложнения включают увеличение МТ, остеопороз, задержку полового созревания, задержку роста, акне, непереносимость глюкозы и катаракту. Учитывая улучшение на фоне стероидов двигательных способностей, а также потенциально состояние в легочном, ортопедическом и сердечном здоровье, они рекомендуются для детей с МДД.
Экзондис 51 (этеплирсен) — это экзон 51, пропускающий олигонуклеотидный подход, который связывает РНК и пропускает дефектный экзон, восстанавливая рамку считывания, тем самым производя более короткий, но потенциально функциональный дистрофиновый белок. Это относится только к пациентам с мутациями, поддающимися этой репарации (13% пациентов). Его вводят в/в еженедельно. Аталурен позволяет считывать преждевременные стоп-кодоны (10-15% пациентов) из нонсенс-мутации, приводящей к выработке функционального дистрофина. Он может иметь преимущества у пациентов на определенном уровне прогрессирования заболевания.
Дополнительные подходы к пропуску экзонов и стратегии замены генов в настоящее время проходят клинические испытания.
Кардиологическая помощь первоначально включает иАПФ, блокаторы рецепторов ангиотензина или бета-блокаторы. Др. используемые ЛП включают антагонисты альдостерона [напр., спиронолактон (Альдактон) или эплеренон]. Оптимальное время для начала лечения сердечными ЛП постоянно изучается; хотя большинство исследователей классически инициировали сердечное лечение в момент снижения ФВ ЛЖ до <55%, некоторые центры выступают за начало лечения до обнаружения аномалий ЭхоКГ. Это основано на более поздних данных, показывающих MPT-изменения в сердце, предшествующие аномалиям ЭхоКГ, и потенциально кардиопротекторный эффект таких ЛП, как иАПФ. Из-за потенциального риска гиперкалиемических или злокачественных реакций гипертермии на анестезию рекомендуется избегать таких ЛС, как ингаляционные анестетики или миорелаксанты.
Легочные инфекции следует немедленно лечить. Пациентам следует избегать контакта с детьми, у которых имеются явные респираторные или другие инфекционные заболевания. Показаны прививки от вируса гриппа и др. плановые прививки. При подозрении на нарушение дыхания во сне дети должны пройти исследования сна и рассмотреть возможность применения BiPAP. Дополнительные устройства, такие как устройство для облегчения кашля, правильного всасывания и ингаляции, могут помочь с орошением ДП, когда у детей развивается слабость при кашле.
Важно сохранение хорошего пищевого состояния. МДД не является витаминно-дефицитным заболеванием, и следует избегать чрезмерных доз витаминов. Адекватное потребление кальция важно для минимизации остеопороза у мальчиков, прикованных к инвалидному креслу. Однако из-за хронического применения ГКС в сочетании с потерей способности к передвижению мальчики с МДД подвергаются более высокому риску остеопении и остеопороза, что повышает риск переломов даже при незначительных травмах или падениях. Сканирование DEXA должно проводиться у мальчиков с МДД, и уровень витамина D должен быть оптимизирован.
Некоторым пациентам с низкой плотностью костной ткани может потребоваться дополнительная терапия, такая как памидроновая кислота (Памидронат медак). Из-за снижения энергозатрат и применения ГКС неходячие дети склонны чрезмерно питаться и набирать избыточную МТ. Ожирение делает пациента с миопатией еще менее функциональным, потому что часть ограниченной резервной мышечной силы рассеивается при увеличении избыточной подкожной жировой ткани. Могут потребоваться диетические ограничения с наблюдением.
Физиотерапия задерживает, но не всегда предотвращает контрактуры. Иногда при контрактурах действительно полезна функциональная реабилитация. Если контрактуры препятствуют разгибанию локтя на >90 градусов и мышцы верхней конечности уже недостаточно сильны, чтобы преодолеть силу тяжести, контрактуры локтя функционально полезны для фиксации руки, и позволяют пациенту есть и писать. Хирургическая коррекция контрактуры локтевого сустава может быть технически осуществима, но результат бывает пагубным. Методы растяжения и фиксации могут быть полезны в зависимости от локализации контрактуры и степени ее тяжести.
Хирургические вмешательства следует рассматривать с осторожностью и при участии невролога, физиотерапевта и/или специалистов по физической медицине и реабилитации, участвующих в уходе за ребенком. Физиотерапия мало способствует укреплению мышц, потому что пациенты обычно уже используют весь свой резерв для ежедневного функционирования, и физические упражнения не могут еще больше укрепить задействованные мышцы. Чрезмерные ФН действительно могут ускорить процесс дегенерации мышечных волокон.
Особую бдительность следует проявлять при наблюдении за прогрессирующим сколиозом, который должен лечиться на ранних стадиях ортопедами с использованием бандажей или корсетов, а иногда и хирургами. Сколиоз часто прогрессирует быстрее, как только пациент теряет способность к самостоятельному передвижению.