Эпизодическая, обратимая слабость или паралич, известный как периодический паралич, связан с преходящими изменениями уровня калия в сыворотке крови, как правило, это гипокалиемия, но иногда гиперкалиемия.
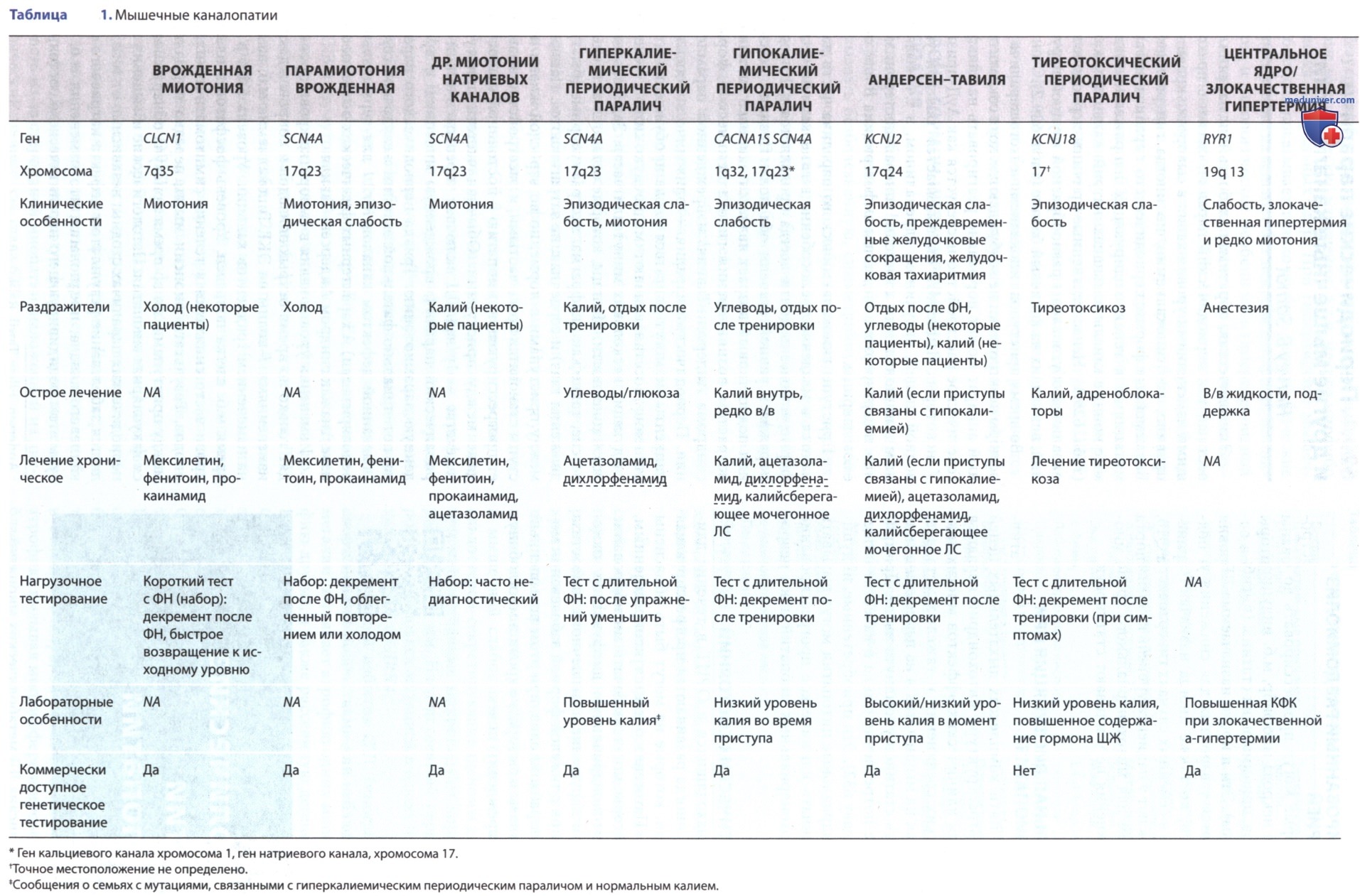
Все семейные формы периодического паралича вызваны мутациями в генах, кодирующих напряженно-зависимые ионные каналы в мышцах: натрий, кальций и калий (табл. 1). Ненаследственные причины периодических параличей обусловлены разнообразной группой нарушений, влияющих на калиевый баланс (табл. 2).
Во время приступов гипокалиемического паралича миофибры электрически не возбуждаются, хотя сократительный аппарат может нормально реагировать на кальций. Генетическое расстройство наследуется как АуД-признак. У некоторых пациентов он вызывается тяжелой углеводной пищей, инсулином, адреналином, в т.ч. вызванным эмоциональным стрессом, гиперальдостеронизмом или гипертиреозом, введением амфотерицина В или приемом лакрицы.
Приступы гипокалиемического паралича часто начинаются в младенчестве, особенно в гиперкалиемической форме, и болезнь почти всегда проявляется к 10 годам, поражая в равной степени оба пола. Поздний детский или подростковый возраст является наиболее типичным возрастом возникновения гипокалиемической формы, синдрома Андерсен-Тавиля (Andersen, Ellen; Tawil, Rabi) и врожденной парамиотонии.
Периодический паралич — эпизодическое явление; пациенты не могут двигаться после пробуждения и постепенно восстанавливают мышечную силу в течение следующих нескольких минут или часов. Задействованы все 4 конечности. Мышцы, которые остаются активными во сне, такие как диафрагма, экстраокулярные (быстрые движения глаз) и сердечная, не затрагиваются.
Пациенты между приступами в норме, но во взрослой жизни приступы становятся более частыми, и расстройство вызывает прогрессирующую миопатию с постоянной слабостью даже между приступами. Обычная частота приступов в детстве — 1 р/нед. ДД включает тиреотоксический периодический паралич, врожденную миотонию и врожденную парамиотонию. Триада периодического паралича, потенциально фатальной эктопии желудочков сердца (вызванной дефектом каналов Kir2.1 для терминальной реполяризации) и характерных физических особенностей известна как синдром Андерсен-Тавиля.
Изменения уровня калия в сыворотке крови происходят только во время острых эпизодов и сопровождаются изменениями Т-волны на ЭКГ. Гипокалиемия м.б. вызвана изменением градиентов кальция. Уровень КФК в это время м.б. слегка повышен.
Уровень фосфатов в плазме крови часто снижается в течение симптоматических периодов. Результаты биопсии мышц нередко нормальны между приступами, но во время приступа обнаруживается вакуолярная миопатия. Патологические изменения при периодических параличах сходны, независимо от того, является заболевание результатом дефекта натриевого или калиевого канала, предполагая, что изменения м.б. результатом рецидивирующего паралитического состояния, а не специфической канальной патологией.
Вакуоли представляют собой расширенные саркоплазматические ретикулумы и инвагинации внеклеточного пространства в цитоплазму, и они м.б. заполнены гликогеном. Однако биопсия мышц не является необходимой для диагностики периодического паралича. Гипогликемия не возникает. Локусы для большинства периодических параличей были продемонстрированы и гены, по крайней мере, частично охарактеризованы, но многие пациенты с тем же клиническим фенотипом не проявляют мутаций в идентифицированных генах.
а) Лечение. Паралитические приступы гипокалиемического периодического паралича лучше всего лечить приемом калия внутрь или из фруктовых соков, содержащих калий. Низкое потребление натрия и введение ацетазоламида, 5 мг/кг/сут Q8-12H в качестве начальной дозы, часто эффективны для прекращения приступов или, по крайней мере, снижения их частоты и тяжести. Дихлорфенамид, ингибитор карбоангидразы, одобрен для лечения первичных гипокалиемических и гиперкалиемических синдромов периодического паралича у взрослых.
ЛП снижал частоту, с небольшими побочными эффектами (парестезии, спутанность сознания, дисгевзия). Ацетазоламид также был использован off label для этих состояний.
б) Другие мышечные каналопатии. Нарушения ионных каналов, отличные от хорошо известных калиевых каналопатий (см. табл. 1). Редкая тяжелая неонатальная миотония вторична по отношению к мутации гена SCN4A натриевого канала с напряжением; она не связана с неонатальной миотонической дистрофией, врожденной миотонией или инфантильной миофибриллярной миопатией. Этот же ген также отвечает за тяжелый неонатальный эпизодический ларингоспазм.
Мексилетин — эффективное ЛС для лечения миотонии, но долгосрочный прогноз остается неблагоприятным, со смертью к 2 годам. Блокаторы натриевых каналов, такие как карбамазепин, фенитоин и прокаинамид, являются альтернативой.
Нейромиотония, непрерывная мышечная активность нейрогенного происхождения, м.б. вызвана мутациями в генах, кодирующих АТл против калиевых каналов, но встречается редко в детском возрасте. Болезнь Шварца-Ямпеля (Schwartz, Oscar; Jampel, Robert Steven), возникающая в результате АуР-наследования, включает сильную мышечную ригидность, миотонию, блефароспазм и хондроплазию.
Она становится симптоматической в первый год жизни и медленно прогрессирует до середины подросткового возраста, после чего стабилизируется. Она больше не считается вариантом миотонической дистрофии и вызвана мутацией в гене HSPG2, который кодирует перлекан, основной гепаринсульфатный протеогликан базальных мембран. М.б. полезны блокаторы натриевых каналов.