Спинальная мышечная атрофия (СМА) — это дегенеративное заболевание двигательных нейронов, которое начинается в эмбриональной жизни и продолжает прогрессировать в младенчестве и детстве. Среди АуР-расстройств в детском возрасте СМА является наиболее распространенной причиной младенческой смертности и уступает по распространенности при рождении только муковисцидозу. По оценкам, частота СМА у новорожденных составляет 1:6000-10 000, при этом частота носительства составляет примерно 1:40-1:60.
Это клинически гетерогенное панэтническое расстройство. СМА вызывается гомозиготной делецией в гене выживания моторного нейрона 1 (SMN1) на хромосоме 5q13. Описаны нечастые семьи с АуД-наследованием, а также редкая Х-сцепленная рецессивная форма. Существует также отдельная группа клинически и генетически гетерогенных форм не-5q СМА.
Патологический признак СМА — прогрессирующая денервация мышц. Это частично компенсируется реиннервацией из соседней двигательной единицы, но т.о. создаются гигантские двигательные единицы с последующей атрофией мышечных волокон, когда в конечном итоге вовлекается реиннервирующий двигательный нейрон.
Двигательные нейроны III, IV и VI ЧМН к экстраокулярным мышцам, а также нейроны крестцового отдела спинного мозга, иннервирующие поперечнополосатую мышцу уретрального и анального сфинктеров, избирательно сохраняются. Верхние двигательные нейроны (пирамидные нейроны 5 слоя в коре ГМ) также остаются нормальными.
СМА клинически классифицируется на тяжелую инфантильную форму, также известную как болезнь Верднига-Гофмана или СМА типа I; позднюю инфантильную и более медленно прогрессирующую форму, СМА типа II; более хроническую или ювенильную форму, болезнь Кугельберга-Веландера или СМА типа III; и взрослую форму (СМА типа IV).
Тяжелая форма плода, которая обычно приводит к летальному исходу в перинатальном периоде, описана как СМА типа 0, с дегенерацией двигательных нейронов, проявляющейся в спинном мозге уже в середине беременности. Эти различия типов основаны на возрасте начала заболевания, степени выраженности слабости, достижении максимального двигательного рубежа и клиническом течении (табл. 7).
Некоторые пациенты являются переходными между типами I и II или между типами II и III с точки зрения клинической функции. Следует отметить, что область гена SMN содержит центромерную копию, содержащую ген выживания моторных нейронов 2 (SMN2). Хотя существует определенная корреляция между тяжестью заболевания, возрастом начала заболевания и числом копий SMN2, считается, что фенотип СМА охватывает широкий континуум без четкого разграничения подтипов.
Мышечная биопсия не различает типы I и II, хотя тип III показывает более взрослый, чем перинатальный, паттерн денервации и реиннервации. Тип 0 может показывать биопсийные находки, более похожие на признаки миотубулярной миопатии из-за задержки созревания; рассеянные миотрубки и др. незрелые фетальные волокна имеются в биопсиях мышц пациентов с типами I и II, но они не преобладают.
Вегетативные моторные нейроны как СНС, так и парасимпатической НС страдают, но обычно не проявляются клинически до поздних стадий. Вегетативный дефицит может вовлекать детрузорную мышцу мочевого пузыря или гладкую мышцу уретрального и анального сфинктеров во всех трех формах СМА. У некоторых пациентов с СМА I типа и РДС может наблюдаться тяжелая вегетативная дисрегуляция с дисавтономией и сердечно-сосудистым коллапсом, приводящим к смерти или тяжелому ишемическому повреждению ГМ. ДД приведена в табл. 8.
а) Этиология. Причина СМА генетическая, как АуР-менделевский признак. По-видимому, это патологическое продолжение процесса запрограммированной клеточной смерти (апоптоза), который является нормальным в эмбриональной жизни. Избыток моторных нейробластов и др. нейронов образуется из примитивной нейроэктодермы, но только около половины выживают и созревают, чтобы стать нейронами; избыток клеток имеет ограниченный жизненный цикл и дегенерирует.
Если процесс, который останавливает физиол. гибель клеток, не вмешивается на определенной стадии, гибель нейронов может продолжаться в конце жизни плода и в постнатальном периоде. Ген SMN задерживает апоптоз моторных нейробластов. В отличие от большинства генов, которые очень консервативны в эволюции, SMN является уникальным геном млекопитающих. Дополнительная функция SMN, как центральная, так и периферическая, заключается в транспортировке РНК-связывающих белков в аксональный ростовой конус, чтобы обеспечить достаточное количество белок-кодирующих транскриптов, необходимых для подвижности ростового конуса, как во время в/утробного развития, так и при постнатальном синаптическом ремоделировании.
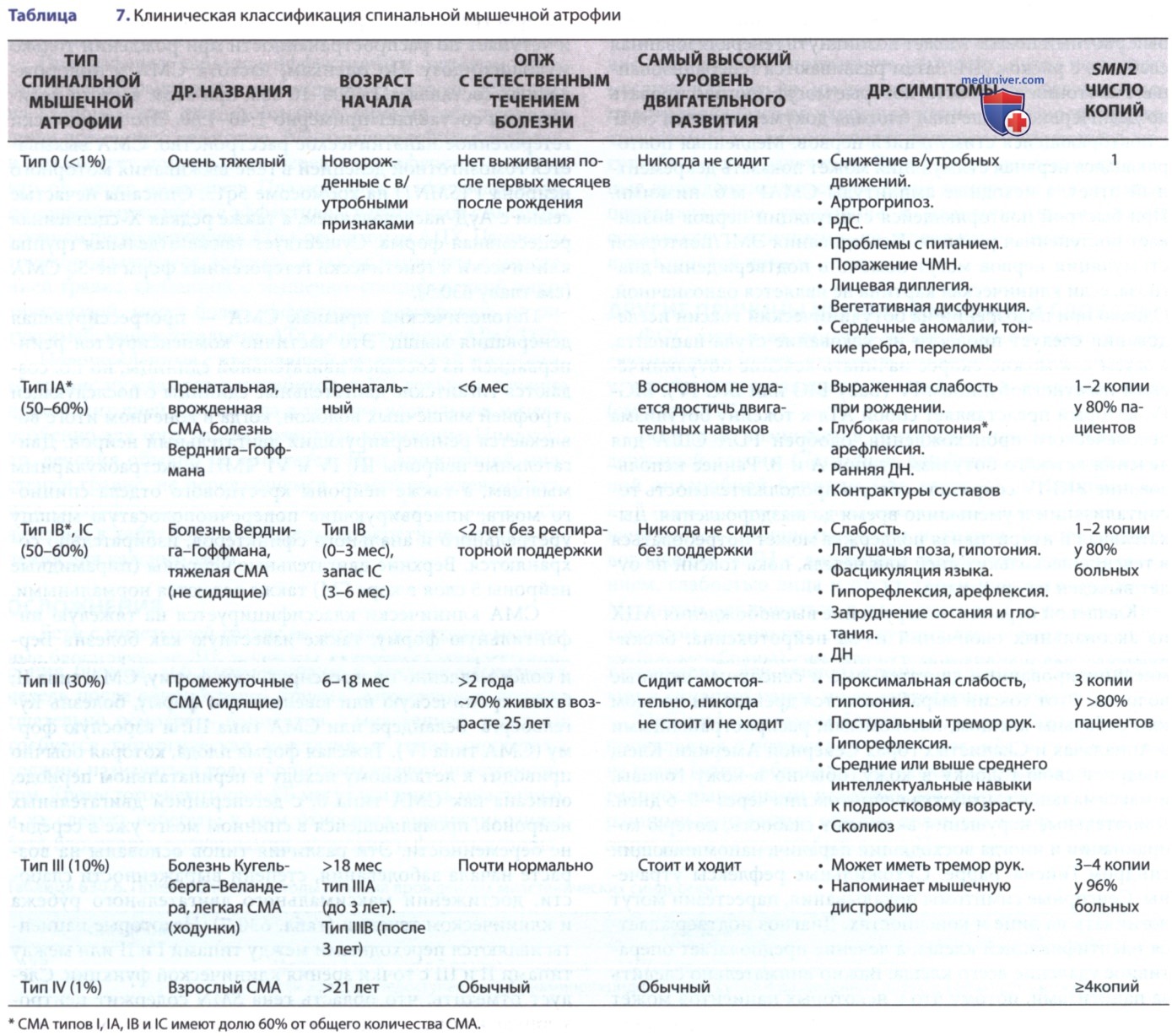
б) Клинические проявления и течение. Кардинальные особенности классического, наиболее распространенного фенотипа СМА I типа можно суммировать как проявление до 6 мес с выраженной гипотонией (рис. 1); симметричная генерализованная мышечная слабость, поражающая нижние конечности больше, чем верхние, проксимальные больше, чем дистальные; поза «лягушачьих лапок»; отсутствие глубоких сухожильных рефлексов; фасцикуляции языка; избирательное вовлечение осевых и межреберных мышц, но щадящее диафрагму.
Рисунок 1. Спинальная мышечная атрофия I типа (болезнь Верднига-Гоффмана): клинические проявления слабости конечностей и осевой мускулатуры у 4-мес ребенка с выраженной слабостью и гипотонией. При вертикальном подвешивании (A) обратите внимание на свисающие нижние конечности с отсутствием сгибания бедер, тенденцию верхних конечностей проскальзывать через руки экзаменатора и отсутствие сгибания шеи с последующим отставанием головы. Когда испытуемый лежит на спине, обратите внимание на положение ног в виде позы «лягушачьей лапки» и отсутствие тягового отклика (B), отставание головы (C) при попытках испытуемого подтянуть ребенка в сидячее положение.
СМА входит в список ДД «синдрома вялого младенца». Из-за вовлечения межреберных дыхательных мышц наблюдается типичный парадоксальный рисунок брюшного дыхания, колоколообразная ГК и слабый кашель. Младенцы вялые, почти не двигаются, не в состоянии преодолеть гравитацию и не контролируют позу голову. Эти дети редко достигают улучшения двигательной функции и приобретают двигательные навыки развития. В отличие от их мышечной слабости и вялости, младенцы с СМА типа I имеют живое и яркое выражение с сохраненными когнитивными функциями. В начале нет вовлечения лицевых и экстраокулярных мышц, хотя слабость мышц лица возникает на более поздних стадиях заболевания.
СМА типа I сама по себе неоднородна. По крайней мере три клинические подгруппы могут быть определены как (1) выраженная слабость с рождения или в неонатальном периоде; контроль позы головы никогда не достигается; (2) представление после неонатального периода, в течение первых 2 мес; контроль позы головы никогда не достигается; и (3) начало после неонатального периода, но контроль позы головы достигается, и некоторые из младенцев могут получить способность сидеть с поддержкой. У этой группы пациентов с СМА типа I может быть целый ряд клинических проявлений и этапов респираторного поражения, а также трудностей с глотанием и сосанием.
У детей с СМА I типа ДН развивается в течение первых 2 лет жизни, без дыхательной и питательной поддержки они обычно не доживают до своего второго дня рождения. Мультидисциплинарный подход (респираторные, желудочно-кишечные и ортопедические вмешательства) в сочетании с неинвазивной вентиляционной поддержкой и энтеральным питанием изменили естественное течение заболевания на протяжении многих лет.
На сегодняшний день медиана времени до смерти или полной неинвазивной вентиляции легких (>16 ч/сут) составляет 13,5 мес при улучшенной дыхательной поддержке и помощи с питанием. Младенцы с симптомами пренатально или при рождении классифицируются как имеющие редкий фенотип, СМА тип 0 (<1%); они имеют выраженную мышечную слабость, РДС, проблемы с питанием и поражение ЧМН. Врожденные контрактуры, начиная от простой косолапости и заканчивая генерализованным артрогрипозом, встречаются у 10% новорожденных с тяжелыми поражениями.
Существует ощущение уменьшения в/утробных движений у матери, и эти младенцы обычно умирают в течение первых месяцев жизни. Хотя двигательные нейроны являются в основном пораженной тканью при СМА, др. ткани, включая ткани ГМ, ССС и даже сенсорных нервов, также могут вносить свой вклад в общий фенотип, особенно при самых тяжелых формах заболевания. Ранние стадии развития ВПС, описанные у тяжелых пациентов с СМА, как правило, несущих одну копию SMN2, включают ДМПП, расширенный ПЖ, ДМЖП и гипопластический синдром левого сердца. Эти пациенты склонны к возможному поражению вегетативной НС, что может привести к аритмии и внезапной смерти.
Васкулопатия может быть еще одним редким проявлением, и изъязвление и некроз пальцев рук и ног также были описаны у двух тяжелых пациентов со СМА I типа.
При СМА II типа младенцы обычно способны сосать и глотать, а дыхание адекватное в раннем младенчестве. Для этой формы достаточно характерна задержка развития грубых двигательных навыков или стагнация двигательного развития в возрасте от 6 до 18 мес. Проксимальная мышечная слабость более заметна в нижних конечностях по сравнению с верхними. Пациенты могут сидеть без поддержки, но не могут самостоятельно ходить. Эти дети имеют прогрессирующую мышечную слабость, но многие доживают до школьных лет или чуть дольше, хотя они прикованы к электрической инвалидной коляске и являются тяжелыми инвалидами.
Носовая речь и проблемы с глотанием развиваются позже. Респираторные осложнения менее тяжелые и развиваются позже по ходу заболевания. Сколиоз становится серьезным осложнением у многих пациентов с длительной выживаемостью. ГЭР может привести к недостаточному питанию или аспирации с острой ОДП или пневмонией.
Болезнь Кугельберга-Веландера — самая легкая СМА (тип III), и пациенты могут казаться нормальными в младенчестве. Прогрессирующая мышечная слабость имеет проксимальное распространение, особенно в мышцах плечевого пояса, после 18 мес у них развивается вариабельное течение слабости проксимальных мышц. Может произойти переход в СМА II типа, и в какой-то момент в течении заболевания может произойти потеря способности к передвижению. Симптомы бульбарной мышечной слабости встречаются редко. Пациенты с этой формой СМА могут иметь мышечную гипертрофию, а не атрофию, и ее легко спутать с мышечной дистрофией.
Продолжительность жизни может продлиться и до взрослого периода. Фасцикуляции являются специфическим клиническим признаком денервации мышц. У худых детей они могут быть видны в дельтовидной и двуглавой мышцах плеча и иногда в четырехглавой мышце бедра, но непрерывные, непроизвольные, червеобразные движения бывают замаскированы толстым слоем ПЖК. Фасцикуляции лучше всего наблюдаются на языке, где почти нет подкожной соединительной ткани, отделяющей мышечный слой от эпителия.
Если внутренние языковые мышцы сокращены, как при плаче или когда язык высовывается, фасцикуляции труднее увидеть, чем когда язык расслаблен. Судороги и миалгии аппендикулярных и аксиальных мышц встречаются часто, особенно на более поздних стадиях, и м.б. проблемы с мочеиспусканием, хотя подростки могут их скрыть, если врач непосредственно не спросит об этом.
В вытянутых пальцах детей со СМА часто наблюдается характерный тремор (полимини-миоклонус) вследствие фасцикуляций и слабости. Его не следует путать с мозжечковым тремором.
Взрослый фенотип заболевания — СМА IV типа, который характеризуется легкой мышечной слабостью с началом обычно во втором или третьем десятилетии жизни.
Может наблюдаться в/семейная вариабельность в клинической выраженности заболевания.
Интеллект в норме, и дети часто кажутся умнее своих здоровых сверстников, потому что усилия, которые они не могут вложить в физическую деятельность, направляются на интеллектуальное развитие, и они часто поддаются воздействию взрослых больше, чем подростков из-за социальных последствий болезни. Прогрессирующее ухудшение осанки и высокий риск падения и перелома длинных костей или таза в конечном итоге требуют использования инвалидной коляски; электрическая инвалидная коляска часто необходима, потому что слабость верхних конечностей не позволяет пациенту вручную толкать колеса.
Прогрессирующий сколиоз является еще одним серьезным осложнением и может оказывать дальнейшее неблагоприятное воздействие на дыхание.
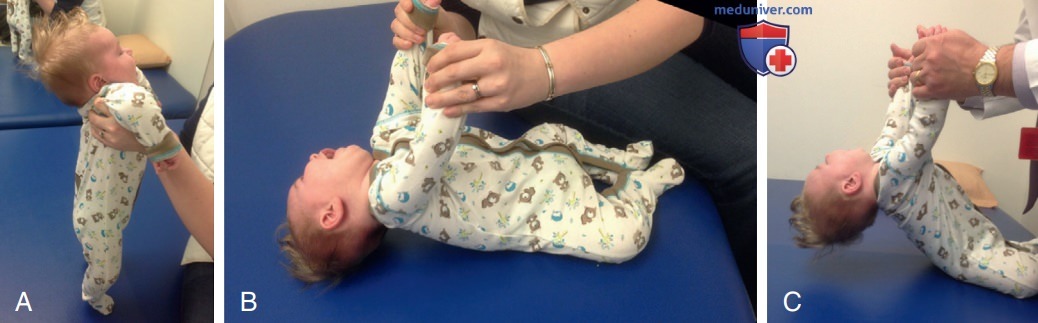
в) Лабораторные результаты. Уровень креатинкиназы в сыворотке крови может быть нормальным, но чаще слегка повышен (до 2-4 раз), обычно не более чем в 10 раз превышает нормальный верхний предел. РОГК при раннем начале заболевания может выявлять истонченные ребра. ЭКГ может служить простым и практичным инструментом у пациентов с СМА для демонстрации исходного тремора как артефакта, представляющего мышечные фибрилляции, более заметные на II отведении (рис. 2). Хотя это наблюдается в основном при заболеваниях периферических двигательных нейронов, включая полиомиелит, распознавание этого паттерна ЭКГ может предотвратить дальнейшие электрофизиол. тесты (ЭМГ и исследования нервной проводимости) у пациентов с СМА.
Рисунок 2. Стандартная 12-канальная электрокардиография (25 мм/с, 10 мВ/мм, диагностический фильтр 0,05-150 Гц) показала диффузную фибрилляцию соматических мышц, исходный тремор, более выраженный на отведении II
Электрофизиол. исследования (ЭМГ и исследования нервной проводимости) должны быть зарезервированы для отдельных атипичных пациентов. Результаты исследований проводимости двигательных нервов нормальны, за исключением умеренного замедления в терминальных стадиях заболевания, что является важным признаком, отличающим СМА от периферической невропатии. ЭМГ показывает потенциалы фибрилляции и др. признаки денервации мышц. Нет необходимости в биопсии мышц, которая показывает нейрогенный паттерн с групповой атрофией при всех формах СМА.
г) Диагноз. Простейшим, наиболее точным диагностическим тестом первого этапа у пациента с клиническим подозрением на СМА и нормальным и/или умеренно повышенным уровнем креатинкиназы в сыворотке крови является молекулярно-генетический маркер гомозиготной делеции в SMN1 (табл. 9). Текущий «золотой стандарт» — тестирование делеции/мутации SMN1 и номера копии SMN2 с минимальным стандартом тестирования делеции SMN1. Отсутствие SMN1 экзона 7 (с делецией экзона 8 или без нее) подтверждает диагноз СМА. Генетический тест на СМА ЧС (95/~100%) (см. табл. 9).
Тесты ПЦР в реальном времени или мультиплексная зависимая от лигирования амплификация зонда дают быстрые и надежные номера копий генов SMN1. Полуколичественные анализы повышают диагностическую чувствительность до 98%. В соответствии с разл. сценариями, напр., если у пациента есть одна копия SMN1, кодирующая область второго неделетированного аллеля должна быть секвенирована для идентификации второй причинной мутации, включая точечные мутации, вставки и делеции. Следует отметить, что у 30% пациентов с клинической картиной мутации не обнаруживаются в кодирующей области SMN1/SMN2, что чаще встречается у пациентов с СМА III типа.
Прямое секвенирование гена также рекомендуется пациентам с клиническим диагнозом, двумя копиями SMN1 и кровным происхождением. Мышечная биопсия была диагностическим тестом до того, как генетический маркер из образцов крови стал доступен, и мышечная биопсия теперь используется более избирательно у пациентов с сомнительными или «-» генетическими результатами.
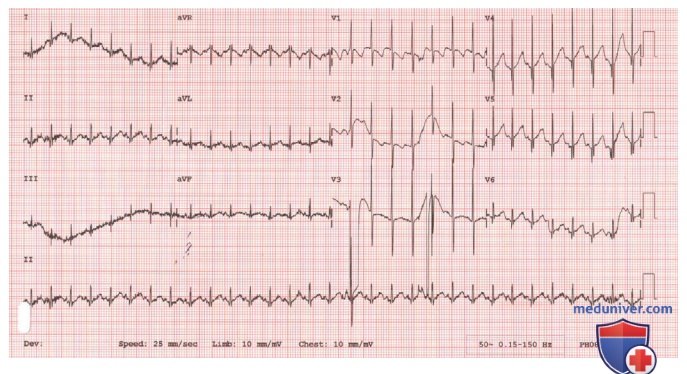
Биопсия мышц в младенчестве выявляет характерный паттерн перинатальной денервации, который не похож на паттерн зрелой мышцы. Группы гигантских волокон I типа смешаны с пучками сильно атрофированных волокон обоих гистохимических типов (рис. 3). Также демонстрируются рассеянные незрелые миофибры, напоминающие миотрубки. В ювенильной СМА картина может быть более похожа на взрослую мышцу, которая прошла много циклов денервации и реиннервации. Нейрогенные изменения в мышцах также могут быть продемонстрированы с помощью ЭМГ, но результаты менее четкие, чем при биопсии мышц в младенчестве.
Рисунок 3. Мышечная биопсия новорожденного с инфантильной спинальной мышечной атрофией. В мышечных пучках сильно атрофированных волокон обоих гистохим. типов видны группы гигантских волокон I типа (темноокрашенных). Это характерная картина перинатальной денервации мышц. Миофибриллярная аденозинтрифосфатаза, предварительно инкубированная при pH 4,6 (х400)
Биопсия сурального нерва в настоящее время проводится лишь изредка, но показывает умеренные сенсорные нейропатические изменения, и скорость проводимости сенсорного нерва может быть замедлена; также наблюдается гипертрофия немиелинизированных аксонов. При аутопсии наблюдаются легкие дегенеративные изменения в сенсорных нейронах дорсальных корневых ганглиев и в соматосенсорных ядрах таламуса, но эти изменения клинически не воспринимаются как потеря чувствительности или парестезии. Наиболее выраженными невропатологическими поражениями являются обширная дегенерация нейронов и глиоз в вентральных рогах спинного мозга и моторных ядрах ствола мозга, особенно в подъязычном ядре.
В редких случаях клинические особенности СМА-подобной презентации могут быть признаком митохондриальных заболеваний (мутации SCO2, DGUOK и ТК2). SCO2 кодирует один из белков сборки СОХ, и последние две мутации генов связаны с синдромами истощения митохондриальной ДНК. Неожиданно повышенный уровень креатинкиназы в сыворотке крови в какой-то момент клинического течения у этих пациентов может быть ключом к рассмотрению митохондриального заболевания в ДД. В зависимости от стадии и прогрессирования заболевания биопсия мышц, демонстрирующая рваные красные волокна и ЦОГ-дефицитные волокна, может помочь в ДД.
д) Генетика. Молекулярно-генетическая диагностика с помощью ДНК-зондов в образцах крови, биопсии мышц или тканей ворсинок хориона доступна для диагностики подозрительных случаев и пренатальной диагностики. Большинство случаев наследуются как АуР-признак.
Генетический локус для всех трех распространенных форм СМА находится на хромосоме 5, делеция в локусе 5q11-q13, указывающая на то, что они являются вариантами одного и того же заболевания, а не разных заболеваний. Измененный ген SMN1 имеет молекулярную массу 38 кДа и содержит 8 экзонов, охватывающих 20 кб, а также теломерные и центромерные экзоны, которые отличаются только на 5 bp и продуцируют транскрипт, кодирующий 294 аминокислоты. SMN1 дублируется в высоко гомологичном гене, называемом SMN2, и оба гена транскрибируются. SMN2 остается присутствующим у всех пациентов с СМА, но не может полностью компенсировать дефект SMN1.
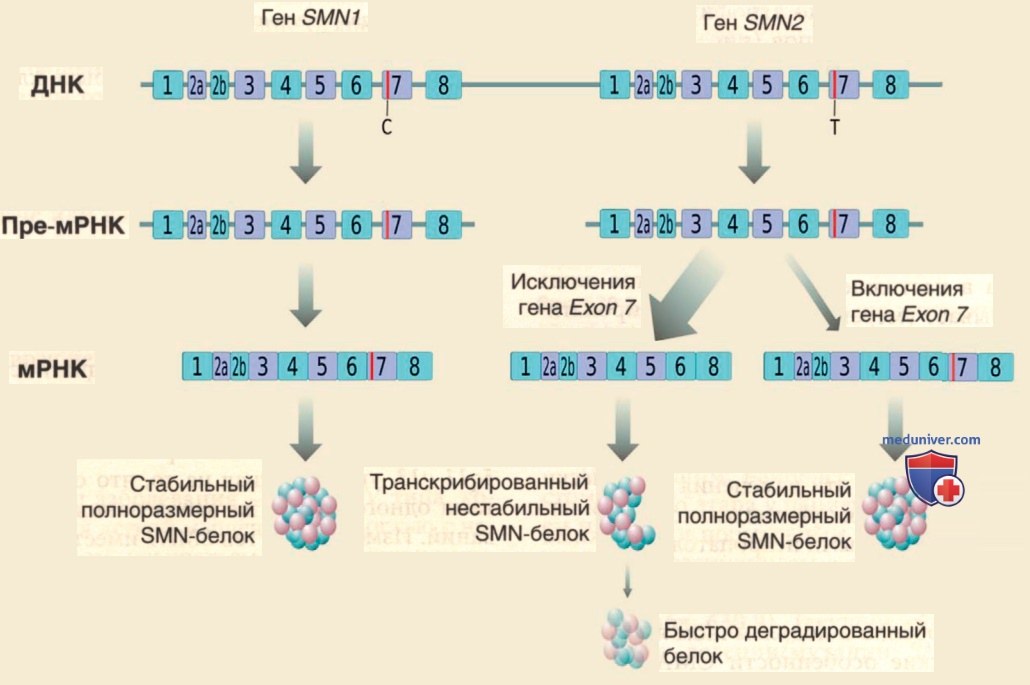
Однако молекулярной основой корреляции между числом копий SMN2 и клинической тяжестью СМА является способность SMN2 кодировать небольшое количество идентичного белка SMN. Критической разницей между SMN1 и SMN2 является переход цитозина (С) в тимин (Т) в экзоне 7 SMN2 (рис. 4).
Рисунок 4. Генетика спинальной мышечной атрофии. У человека белок SMN кодируется генами SMN1 и SMN2. Замена С на Т в экзоне 7 SMN2 трансляционно молчит, но изменяет сплайсинг т.о., что большинство транскриптов SMN2 не имеют экзона 7, а усеченный белок нестабилен. В норме SMN1 производит обильный белок SMN. При спинальной мышечной атрофии гомозиготная мутация SMN1 приводит лишь к небольшому количеству функционального белка SMN, вносимого разл. числами копий SMN2. мРНК 5, мессенджерная РНК; SMN 5, моторный нейрон выживания
Комплекс SMN играет важную роль в образовании малых ядерных рибонуклеопротеинов (snRNPs) путем сборки Sm-белков (отличительного семейства РНК-ассоциированных малых белков) на малые ядерные РНК (snRNAs). Предполагается, что дефицит SMN и снижение способности малых ядерных РНК к сборке вызывают аберрантный сплайсинг или транспорт малых ядерных рибонуклеопротеинов к моторным нейронам. Дисрегуляция генов, участвующих в синаптогенезе и поддержании нервно-мышечных соединений в исследованиях на животных, возможно, объясняет особую уязвимость моторных нейронов.
Вторая точка зрения заключается в том, что, независимо от сборки малых ядерных рибонуклеопротеинов, SMN может играть специфическую для моторных нейронов роль, такую как транспорт мРНК вдоль аксона.
Учитывая длину аксонов, целостность нервно-мышечных соединений и взаимодействие со скелетными мышцами, дефицит белка SMN может быть деструктивным для моторных нейронов. SMN локализуется в ярких точечных структурах, называемых тельцами (близнецами тел Кахаля) в ядре. Он также присутствует в др. клеточных структурах, таких как тела Гольджи, клеточные мембраны и особенно аксонные и ростовые конусы моторных нейронов. Благодаря своей локализации в гранулах рибонуклеопротеинов в нейритах и ростовых колбочках в нейронах SMN модулирует аксональный рост и локализацию рибонуклеиновой кислоты-мессенджера β-актина (мРНК) в ростовых колбочках двигательных нейронов.
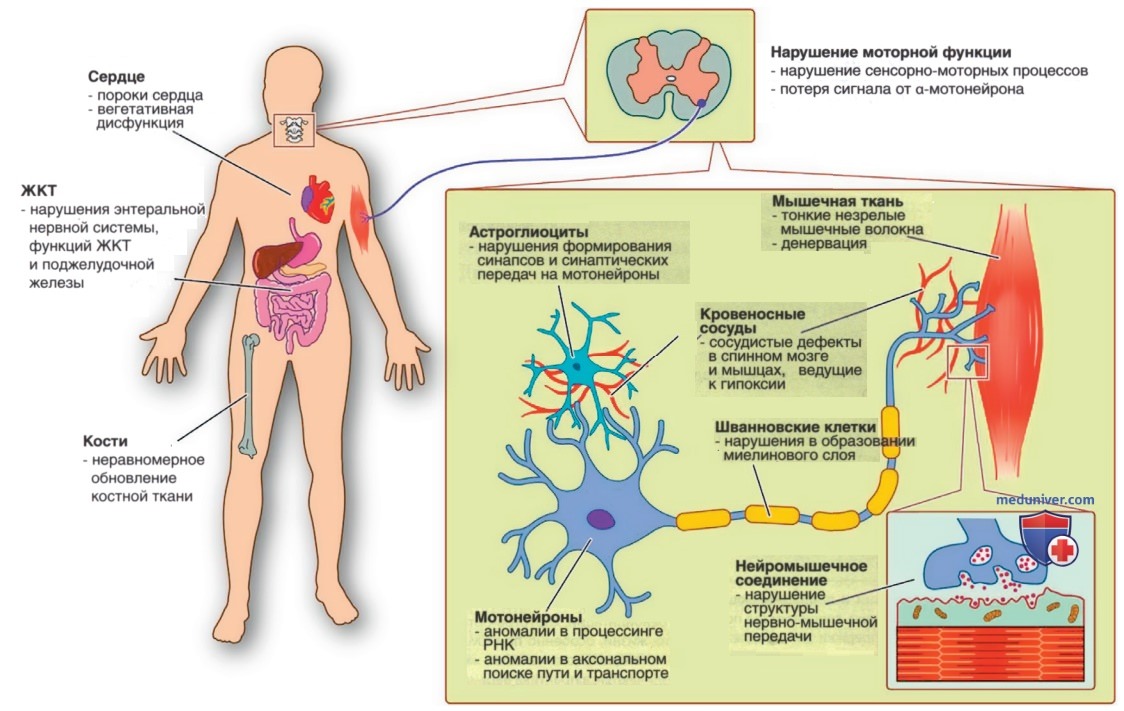
Раннее функциональное нарушение сенсомоторных связей на животных моделях показало, что потеря двигательных нейронов следует за потерей афферентных синапсов с той же временной и топографической картиной, причем изменения происходят сначала в двигательных нейронах, иннервирующих проксимальные мышцы и аксиальные мышцы, а затем дистальные мышцы. Третья точка зрения прямо или косвенно связывает функцию SMN с динамикой актина и актин-зависимыми процессами. Наблюдается расширение спектра функций SMN, включая динамику актина, везикулярный транспорт, трансляцию и транспорт белков, транспорт мРНК, апоптоз и многие др., что отражается на широко распространенных патофизиол. находках, описанных на моделях человека и животных (рис. 5).
Рисунок 5. Патофизиол. данные при спинальной мышечной атрофии. У мышей и людей со спинальной мышечной атрофией выявлены многочисленные функциональные нарушения в двигательных сетях, включая дефекты в астроцитах, шванновских клетках, двигательных нейронах и скелетных мышцах. Ассоциированные с болезнью фенотипы также зарегистрированы в ряде др. органов у мышей со спинальной мышечной атрофией (в некоторых случаях подтвержденных данными от пациентов-людей), включая структурные и функциональные нарушения сердца, дисфункцию желудочно-кишечного тракта и нерегулярное ремоделирование костей. Одним из потенциальных объединяющих факторов может быть дефицит в развитии сосудистой системы при спинальной мышечной атрофии, причем возникающая в результате гипоксия, вероятно, влияет на целый ряд типов клеток
Тяжесть заболевания обратно коррелирует с количеством функционального белка SMN. В этом смысле, помимо числа копий SMN2, которое является основным защитным модификатором, на тяжесть фенотипа могут влиять и др. генетические модификаторы, включая пластин 3 и нейрокальцин. Дефицит питательных в-в, окислительный стресс и гипоксия могут способствовать широко распространенным изменениям сплайсинга, включая SMN2, и влиять на прогрессирование заболевания.
Тестирование носителей с помощью анализа дозы доступно и основано на полуколичественной ПЦР в реальном времени или на мультиплексной зависимой от лигирования амплификации зонда. В этом контексте следует учитывать ограничения молекулярного тестирования, трудности в прогнозировании фенотипа потомства, основанного исключительно на количестве копий SMN2, и влияние на репродуктивное планирование.
Скрининг новорожденных направлен на выявление пресимптомных пациентов с СМА. Разработано извлечение ДНК из пятен крови новорожденных с последующим использованием либо жидкого микробного массива, либо методов ПЦР в реальном времени, которые помогают идентифицировать гомозиготные делеции SMN1. Проблемы при скрининге новорожденных включают неспособность обнаружить носителей гетерозиготных делеций числа копий SMN1 и SMN2.
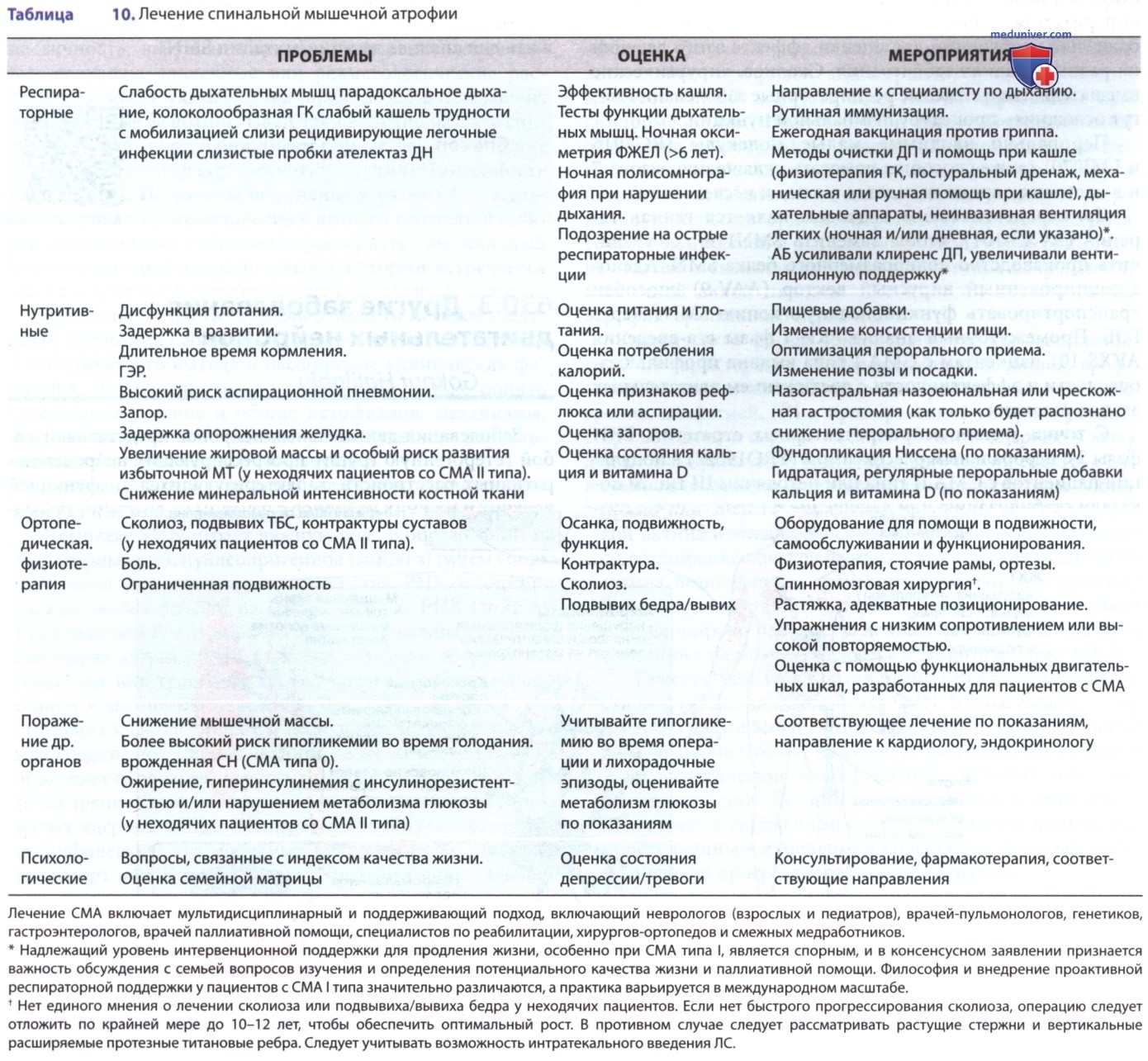
е) Ведение пациентов (лечение). Мультидисциплинарный и поддерживающий подход является ключевым в ведении пациента со СМА. Последующая координация должна осуществляться специалистом по нервно-мышечным расстройствам, и в идеале команда должна включать педиатра и невролога, врачей-пульмонологов, генетиков, гастроэнтерологов, врачей паллиативной помощи, специалистов по реабилитации, хирургов-ортопедов и смежных медработников. Консенсусное заявление о стандарте МП при СМА включает разделы этической и паллиативной помощи. Несмотря на возросшие стандарты и технологические достижения, существует высокая вариабельность с точки зрения вентиляционной поддержки, питательной поддержки и хирургии сколиоза.
С точки зрения достижений в области модифицирующих болезнь методов лечения, которые изменят естественное течение болезни, варианты ухода и лечения должны быть четко обсуждены с семьей и/или пациентами, чтобы определить ожидания, качество жизни и вопросы паллиативной помощи. Поскольку СМА по своей природе является динамичным заболеванием, активный план должен быть введен почти в каждом виде ухода (табл. 10). В целом поддерживающая терапия должна быть направлена на то, чтобы помочь пациенту быть как можно более функционально независимым.
1. Терапевтические достижения. SMN-антисмысловой олигонуклеотид, нусинерсен, вводимый интратекально, одобрен FDA США и Европейским агентством по ЛС для всех типов пациентов со СМА. Он модифицирует сплайсинг SMN2, индуцируя увеличение удержания экзона 7 в пре-мРНК SMN2, что в конечном итоге позволяет получить белковый продукт, подобный SMN1. Исследования фазы 1-3 у пациентов со СМА типа I (0-6 мес) и СМА типа II/III (2-14 лет) показали благоприятную безопасность, переносимость и обнадеживающую клиническую эффективность. Первичная конечная точка была достигнута в каждом исследовании при промежуточном анализе со статистически значимым улучшением двигательного развития.
В продолжающемся открытом клиническом исследовании этот эффект также был протестирован на пресимптомных пациентах со СМА, и до сих пор результаты были благоприятными. Длительное наблюдение необходимо для оценки эффекта этого лечения на разных стадиях заболевания. Сколиоз, хирургические вмешательства и тяжелые респираторные заболевания могут осложнить процедуру люмбальной пункции.
Перорально вводимые малые молекулы (RG7916 и LMI070) также способны помогать включению экзона 7 и в настоящее время находятся в стадии исследования.
Др. терапевтическим подходом является генная терапия (AVXS-101), чтобы заменить SMN1 и т.о. увеличить производство полноразмерного белка SMN. Аденоассоциированный вирусный вектор (AAV-9) способен транспортировать функциональную копию SMN1 через ГЭБ. Промежуточный анализ РКИ I фазы в/в-введения AVXS-101 пациентам с СМА I типа выявил профиль безопасности и эффективности с достижением двигательных этапов разаития.
С точки зрения нейропротекторных стратегий, РКИ фазы 2 с пероральным олесоксимом (TRO19622) в популяции пациентов с СМА II типа или неходячим III типом показали стабилизацию или улучшение по сравнению с плацебо. Хотя первичная конечная точка не была достигнута, олесоксим был безопасен и мог использоваться в комбинации с др. ЛП, нацеленными на др. механизмы заболевания. Роль физических упражнений как нейропротекторной меры также изучается. Текущие клинические испытания также включают быстрый скелетный активатор тропонина (CK2127107), пиридостигмина бромид и 4-аминопиридин для усиления нервной или мышечной функции.
Генетическое консультирование в зависимости от скрининговых тестов носителей или при наличии ранее заболевшего ребенка со СМА может помочь в репродуктивном планировании (пренатальная диагностика или предимплантационная диагностика). Пренатальная диагностика должна быть предложена семьям с индексным пациентом в семье (риск рецидива составляет 25%), а дородовый скрининг путем отбора проб ворсинок хориона между 10-й и 12-й неделями беременности может служить для анализа делеции/мутации SMN1.