Гликогенозы у детей - кратко с точки зрения педиатрии
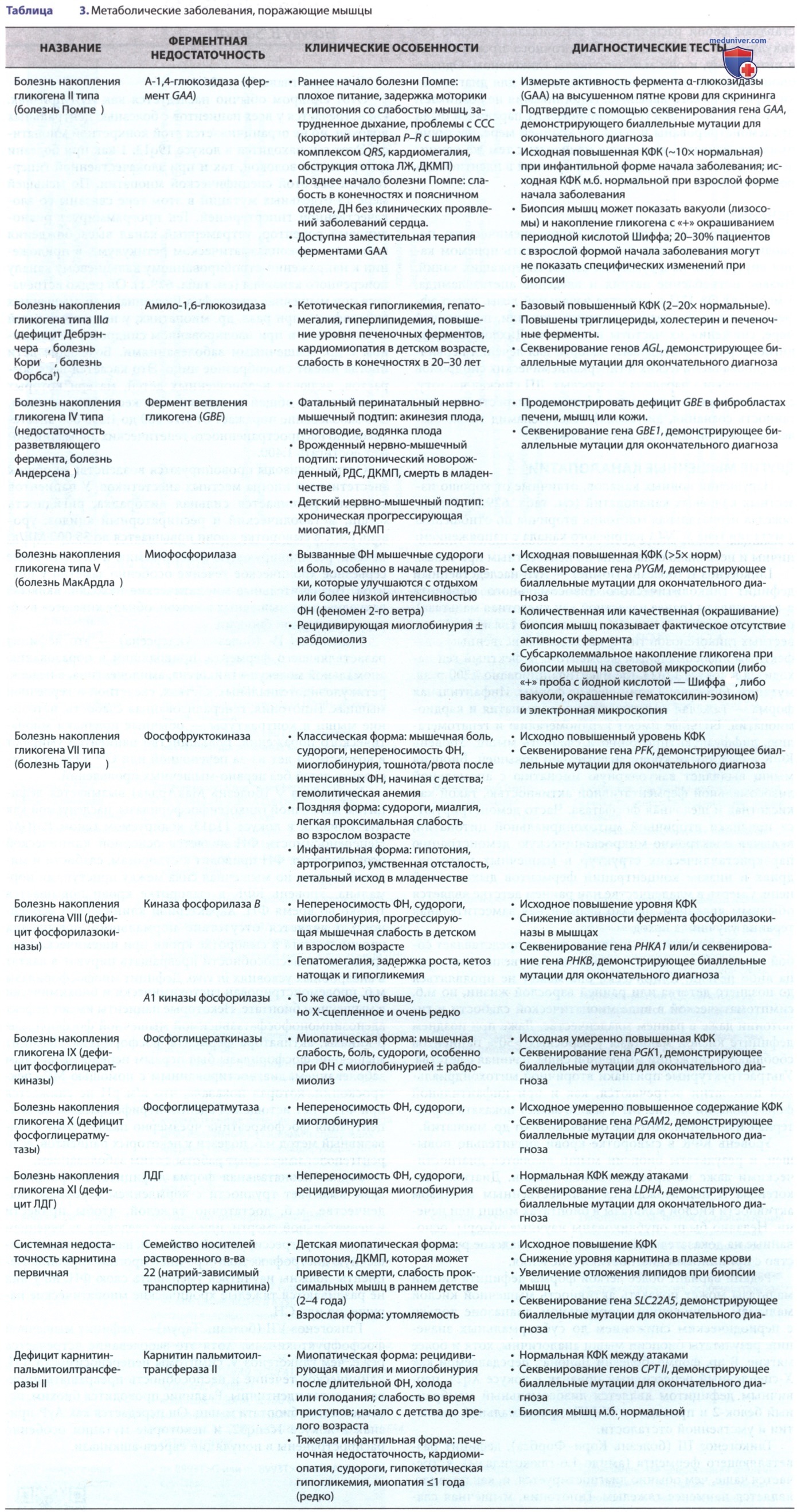
См. табл. 3.
Гликогеноз I (болезнь фон Гирке (Gierke, Edgar Otto Conrad von)) не является истинной миопатией, потому что недостаточный печеночный фермент глюкозо-6-фосфатаза обычно не присутствует в мышцах. Тем не менее дети с этим заболеванием гипотоничны и несколько ослаблены по неизвестным причинам.
Гликогеноз II (болезнь Помпе) — АуР-наследственный дефицит гликолитического лизосомального фермента а-глюкозидазы (ранее известной как кислотная мальтаза), расщепляющего α-1,4 и α-1,6 гликозидные связи.
Из 12 известных гликогенозов тип II является единственным с дефектным лизосомальным ферментом. Дефектный ген находится в локусе 17q23, где идентифицировано >200 разл. мутаций. Описаны 2 клинические формы. Инфантильная форма — тяжелая генерализованная миопатия и кардиомиопатия. Больные имеют кардиомегалию и гепатомегалию, диффузную гипотонию и слабость мышц. Уровень КФК в сыворотке крови значительно повышен.
Биопсия мышц выявляет вакуолярную миопатию с аномальной лизосомальной ферментативной активностью, такой как кислотная и щелочная фосфатаза. Часто демонстрируются признаки вторичной митохондриальной цитопатии, включая электронно-микроскопическую демонстрацию паракристаллических структур в мышечных митохондриях и низкие концентрации ферментов дыхательной цепи. Смерть в младенчестве или раннем детстве является обычным явлением, однако ферментная заместительная терапия улучшила исход.
Поздняя детская или взрослая форма представляет собой гораздо более мягкую миопатию без увеличения сердца либо печени. Клинически она может не проявляться до позднего детства или ранней взрослой жизни, но м.б. симптоматической в виде миопатической слабости и гипотонии даже в раннем младенчестве. Даже при позднем дефиците кислой мальтазы у взрослых >50% пациентов сообщают о снижении мышечной силы, начиная с детства. Ультраструктурные признаки вторичной митохондриальной цитопатии встречаются, как и при инфантильной форме болезни Помпе. МРТ мышц может показать характерные изменения, которые отличаются от др. миопатий.
Уровень КФК в сыворотке крови значительно повышен, и результаты биопсии мышц являются диагностическими даже на пресимптомной стадии. Диагноз гликогеноза II подтверждается количественным анализом активности кислой мальтазы в биоптатах мышц или печени. Недавно были опубликованы научные обзоры, основанные на доказательной медицине, и Канадское руководство с принципами диагностики и лечения.
Редкий вариант более легкой формы дефицита кислой мальтазы может показать активность мышечной кислой мальтазы в сниженном нормальном диапазоне только с периодическим снижением до субнормальных значений; результаты биопсии мышц аналогичны, хотя и более мягкие. В др. форме, болезни Данона (Danon, Moris), передаваемой как Х-сцепленный рецессивный признак в локусе Xq24, первичным дефицитом является лизосомальный мембранный белок-2 и приводит к ГКМП, проксимальной миопатии и умственной отсталости.
Гликогеноз III (болезнь Кори-Форбса), дефицит разветвляющего фермента (амило-1,6-глюкозидазы), встречается чаще, чем обычно диагностируется, и, как правило, является наименее тяжелым. Гипотония, мышечная слабость, гепатомегалия и гипогликемия натощак в младенчестве являются общими, но эти особенности часто разрешаются спонтанно, и у пациентов отсутствуют симптомы в детстве и взрослой жизни. Др. испытывают медленно прогрессирующее истощение дистальных мышц, цирроз печени, рецидивирующую гипогликемию и СН. Это более серьезное хроническое течение особенно заметно у инуитов.
Незначительные миопатические находки, включая вакуолизацию мышечных волокон, обнаруживаются в образце мышечной биопсии.
Гликогеноз IV (болезнь Андерсена) — это дефицит разветвляющего фермента, приводящий к образованию аномальной молекулы гликогена, амилопектина, в печени, ретикулоэндотелиальных клетках, скелетной и сердечной мышцах. Гипотония, генерализованная слабость, истощение мышц и контрактуры — обычные признаки миопатического поражения. Большинство пациентов умирают в возрасте <4 лет из-за печеночной или СН. Описано несколько детей без нервно-мышечных проявлений.
Гликогеноз V (болезнь МакАрдла) вызывается дефицитом мышечной гликогенфосфорилазы, наследуемой как АуР-признак в локусе 11q13, кодируемом геном PMGM. Непереносимость ФН является основной клинической особенностью. ФН приводит к судорогам, слабости и миоглобинурии, но мышечная сила между приступами нормальна. Уровень КФК в сыворотке крови повышается только во время ФН. Характерной клинической особенностью является отсутствие нормального повышения уровня лактата в сыворотке крови при ишемической нагрузке из-за неспособности превращать пируват в лактат в анаэробных условиях in vivo.
Дефицит миофосфорилазы м.б. продемонстрирован гистохимически и биохимически в мышечном биоптате. Некоторые пациенты имеют дефект аденозинмонофосфатзависимой мышечной фосфорилазы β-киназы, активатора фермента фосфорилазы. Дефицит мышечной фосфорилазы был первым нервно-мышечным заболеванием, диагностированным с помощью МР-спектроскопии, которая показала, что в/м pH не снижается при ФН и нет истощения аденозинтрифосфатазы, но концентрация фосфокреатина чрезмерно падает. Этот неинвазивный метод м.б. полезен у некоторых пациентов, если рентгенолог имеет опыт работы с этим заболеванием.
Редкая неонатальная форма дефицита миофосфорилазы вызывает трудности с кормлением в раннем младенчестве, м.б. достаточно тяжелой, чтобы привести к неонатальной смерти, или может следовать за течением медленно прогрессирующей слабости, напоминающей мышечную дистрофию. Долгосрочный прогноз хороший. Пациенты должны научиться уменьшать свою ФН, но у них не развиваются тяжелые хронические миопатические нарушения или СН.
Гликогеноз VII (болезнь Таруи) — дефицит мышечной фосфофруктокиназы. Хотя это заболевание встречается реже, чем гликогеноз V, симптомы непереносимости ФН, клиническое течение и неспособность превращать пируват в лактат идентичны. Различие проводится биохим. исследованием биоптата мышц. Он передается как АуР-признак в локусе lcenq32, и некоторые мутации особенно распространены в популяции евреев-ашкинази.